Introduction
The thyroid gland synthesizes the hormones thyroxine (T4) and triiodothyronine (T3), iodine-containing amino acids that regulate the body’s metabolic rate. Adequate levels of thyroid hormone are necessary in infants for normal development of the CNS, in children for normal skeletal growth and maturation, and in adults for normal function of multiple organ systems. Thyroid dysfunction is one of the most common endocrine disorders encountered in clinical practice. Although abnormally high or low levels of thyroid hormones may be tolerated for long periods of time, usually there are symptoms and signs of overt thyroid dysfunction.
Normal Structure & Function
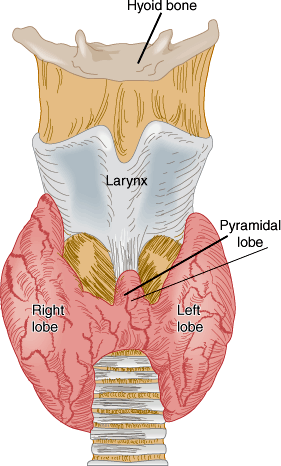
The normal thyroid gland is a firm, reddish brown, smooth gland consisting of two lateral lobes and a connecting central isthmus (Figure 20–1). A pyramidal lobe of variable size may extend upward from the isthmus. The normal weight of the thyroid ranges from 30–40 g. It is surrounded by an adherent fibrous capsule from which multiple fibrous projections extend deeply into its structure, dividing it into many small lobules. The thyroid is highly vascular and has one of the highest rates of blood flow per gram of tissue of any organ.
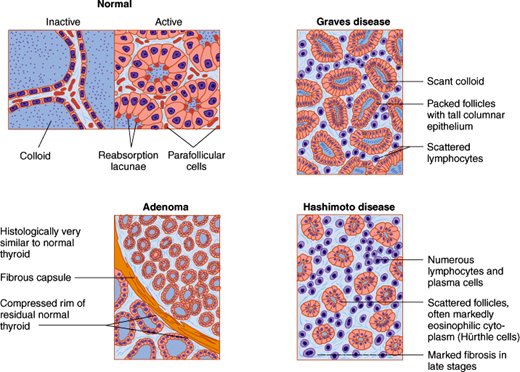
Histologically, the thyroid gland consists of many closely packed acini, called follicles, each surrounded by capillaries and stroma. Each follicle is roughly spherical, lined by a single layer of cuboidal epithelial cells and filled with colloid, a proteinaceous material composed mainly of thyroglobulin and stored thyroid hormones. When the gland is inactive, the follicles are large, the lining cells are flat, and the colloid is abundant. When the gland is active, the follicles are small, the lining cells are cuboidal or columnar, the colloid is scanty, and its edges are scalloped, forming reabsorption lacunae (Figure 20–2). Scattered between follicles are the parafollicular cells (C cells), which secrete calcitonin, a hormone that inhibits bone resorption and lowers the plasma calcium level (see Chapter 17).
Figure 20–2
Normal and abnormal thyroid histology. (Redrawn, with permission, from Barrett KE et al, eds. Ganong’s Review of Medical Physiology, 24th ed. McGraw-Hill, 2012; Chandrasoma P et al, eds. Concise Pathology, 3rd ed. Originally published by Appleton & Lange. Copyright © by The McGraw-Hill Companies, Inc.; Gardner DG et al, eds. Greenspan’s Basic and Clinical Endocrinology, 9th ed. McGraw-Hill, 2011.)
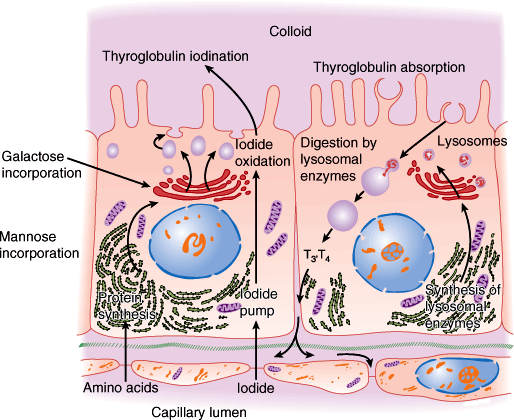
The ultrastructure of a follicular epithelial cell is diagrammed in Figure 20–3. The cells vary in appearance with the degree of gland activity. The follicular cell rests on a basal lamina. The nucleus is round and centrally located. The cytoplasm contains mitochondria, rough endoplasmic reticulum, and ribosomes. The apex has a discrete Golgi apparatus, small secretory granules containing thyroglobulin, and abundant lysosomes and phagosomes. At the apex, the cell membrane is folded into microvilli.
Figure 20–3
Thyroid cell ultrastructure (schematic). The processes of synthesis and iodination of thyroglobulin are shown on the left and its reabsorption and digestion on the right. (Redrawn, with permission, from Junqueira LC et al, eds. Basic Histology, 9th ed. Originally published by Appleton & Lange. Copyright © 1998 by The McGraw-Hill Companies, Inc.)
Thyroid follicular cells have three functions: (1) collect and transport iodine to the colloid; (2) synthesize thyroglobulin, a 660,000-Da glycoprotein made up of two subunits and containing many tyrosine residues, and secrete it into the colloid; and (3) release thyroid hormones from thyroglobulin and secrete them into the circulation. The structures of the two thyroid hormones, T3 and T4, are shown in Figure 20–4. T3 and T4 are synthesized in the colloid by iodination and condensation of tyrosine molecules bound together in thyroglobulin.
For normal thyroid hormone synthesis, an adult requires a minimum daily intake of 150 μg of iodine. In the United States, the average intake is about 500 μg/d. Iodine ingested in food is first converted to iodide, which is absorbed and taken up by the thyroid. The follicular cells transport iodide from the circulation to the colloid (“iodide trapping” or “iodide pump”). The sodium-iodide symporter is a 65-kDa cell membrane protein. This iodide transport is an example of secondary active transport dependent on Na+-K+ adenosine triphosphatase (ATPase) for energy; it is stimulated by thyroid-stimulating hormone (TSH, thyrotropin). At the normal rate of thyroid hormone synthesis, about 120 μg/d of iodide enters the thyroid. About 80 μg/d is secreted in T3 and T4, and the rest diffuses into the extracellular fluid and is excreted in the urine.
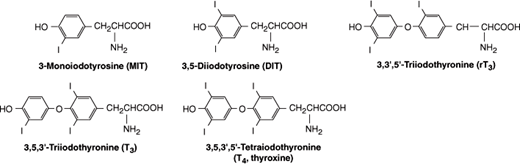
Thyroid hormones are synthesized in the colloid, near the apical cell membrane of the follicular cells. Catalyzed by the enzyme thyroidal peroxidase, iodide in the thyroid cell is oxidized to iodine. The iodine enters the colloid and is rapidly bound at the 3 position (Figure 20–4) to tyrosine molecules attached to thyroglobulin, forming monoiodotyrosine (MIT). MIT is next iodinated at the 5 position, forming diiodotyrosine (DIT). Two DIT molecules then condense in an oxidative process (“coupling reaction”) catalyzed by thyroperoxidase to form one thyroxine (T4) molecule. Some T3 is probably formed within the thyroid gland by condensation of MIT with DIT. A small amount of reverse T3 (rT3) is also formed. Figure 20–4 shows the structures of MIT, DIT, T4, T3, and reverse T3. In the normal thyroid, the average distribution of iodinated compounds is 23% MIT, 33% DIT, 35% T4, 7% T3, and 2% reverse T3.
The thyroid secretes about 80 μg (103 nmol) of T4 and 4 μg (7 nmol) of T3 per day. The folds of the apical cell membrane (lamellipodia) encircle bits of colloid and bring them into the cytoplasm by endocytosis, forming endosomes. This process is accelerated by TSH. The endosomes fuse with lysosomes containing proteases that break peptide bonds between the iodinated residues and thyroglobulin, releasing T4, T3, DIT, and MIT into the cytoplasm. The free T4 and T3 then cross the cell membrane and enter adjacent capillaries. The MIT and DIT are enzymatically degraded in the cell by thyroid deiodinase (iodotyrosine dehalogenase) to iodine and tyrosine, which are reused in colloid synthesis.
The normal plasma level of T4 is approximately 8 μg/dL (103 nmol/L) (range: 5–12 μg/dL or 65–156 nmol/L), and the normal plasma level of T3 is approximately 0.15 μg/dL (2.3 nmol/L) (range: 0.08–0.22 μg/dL or 1.2–3.3 nmol/L). Both hormones are bound to plasma proteins, including albumin, transthyretin (formerly called thyroxine-binding prealbumin [TBPA]), and thyroxine-binding globulin (TBG). The thyroid hormone-binding proteins serve mainly to transport T4 and T3 in the serum and to facilitate uniform distribution of hormones within tissues.
Physiologically, it is the free (unbound) T4 and T3 in plasma that are active and inhibit pituitary secretion of TSH. The free T4 and T3 are in equilibrium with the protein-bound hormones in plasma and tissue and circulate in much lower concentrations. Tissue uptake of the free hormones is proportionate to their plasma concentrations.
Almost all (99.98%) of the circulating T4 is bound to thyroxine-binding globulin (TBG) and other plasma proteins, so that the free T4 level is approximately 2 ng/dL. The biologic half-life of T4 is long (about 6–7 days). Somewhat less T3 (99.8%) is protein bound. Therefore, compared with T4, T3 acts more rapidly and has a shorter half-life (about 30 hours). It is also three to five times more potent on a molar basis.
T4 and T3 are metabolized in the liver, kidneys, and many other tissues by deiodination and by conjugation to glucuronides. Normally, one-third of circulating T4 is converted to T3 by 5′-deiodination, and 45% is converted to the metabolically inert reverse triiodothyronine (rT3) by 5-deiodination. About 87% of circulating T3 derives from peripheral conversion of T4 to T3 and only 13% from thyroid secretion. Both T4 and T3 are conjugated to glucuronides in the liver and excreted into the bile. On passage into the intestine, the conjugates are hydrolyzed, and small amounts of T4 and T3 are reabsorbed (enterohepatic circulation). The rest is excreted in the stool.
Thyroid hormone secretion is stimulated by pituitary thyroid-stimulating hormone (TSH, thyrotropin). Pituitary TSH secretion is, in turn, stimulated by thyrotropin-releasing hormone (TRH), a tripeptide secreted by the hypothalamus that also increases the biologic activity of TSH, by altering its glycosylation.
TSH is a two-subunit glycoprotein containing 211 amino acids. The α-subunit is identical to that of pituitary follicle-stimulating hormone (FSH), luteinizing hormone (LH), and placental human chorionic gonadotropin (hCG). The β-subunit confers the specific binding properties and biologic activity of TSH. The gene encoding the α-subunit is located on chromosome 6, and the gene for the β-subunit is on chromosome 1.
TSH has a biologic half-life of about 60 minutes. The average plasma level of TSH is 2 mU/L (normal range: 0.4–4.8 mU/L). When individuals with autoantibodies, goiter, or a family history of thyroid disease are excluded, the upper limit is somewhat lower, between 2.5–3.0 mU/L. Controversy exists over the effects of advanced age on the normal range for TSH. Several population-based studies have found that the upper limit of normal in healthy older individuals (older than 80 years) might be as high as 7.5 mU/L, but the clinical significance of using age-specific upper limit cut-points remains unclear. Although the phenomenon is not clinically important, normal TSH secretion exhibits a circadian pattern, rising in the afternoon and evening, peaking after midnight, and declining during the day.
Circulating free T4 and T3 inhibit TSH secretion by the pituitary both directly and indirectly by regulating biosynthesis of TRH in the hypothalamus. TSH secretion is inhibited by stress, perhaps via glucocorticoid inhibition of TRH secretion. In infants, but not in adults, TSH secretion is increased by cold and inhibited by warmth. Dopamine and somatostatin also inhibit pituitary secretion of TSH. In animals, there is a pituitary-specific form of the thyroid hormone receptor that may be selectively regulated by thyroid hormone. Figure 20–5 illustrates the hypothalamic-pituitary-thyroid axis and various stimulatory and inhibitory factors.
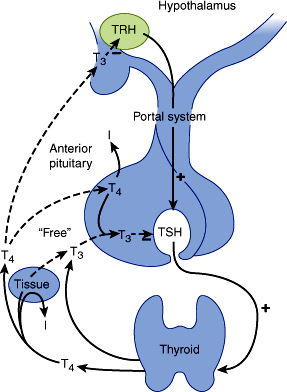
When TSH is secreted or administered, it binds to a specific TSH receptor (TSH-R) in the thyroid cell membrane, activating the GTP-binding (Gs) protein-adenylyl cyclase-cyclic adenosine monophosphate (cAMP) cascade. The increase in intracellular cAMP mediates immediate increases in uptake and transport of iodide, iodination of thyroglobulin, and synthesis of iodotyrosines T3 and T4. Within a few hours, there is an increase in mRNA for thyroglobulin and thyroidal peroxidase, enhanced lysosomal activity, increased secretion of thyroglobulin into colloid, more endocytosis of colloid, and increased secretion of T4 and T3 from the gland. TSH receptor is also expressed in lymphocytes and other tissues, including the pituitary, thymus, kidney, testis, brain, adipocytes, and fibroblasts. TSH-R has also been detected on osteoblast precursors, suggesting that TSH may have a direct effect on bone resorption.
TSH binding to TSH receptor also stimulates membrane phospholipase C, which leads to thyroid cell hypertrophy. With chronic TSH stimulation, the entire gland hypertrophies, increases in vascularity, and becomes a goiter.
TSH receptor has been cloned. It is a single-chain glycoprotein composed of 744 amino acids. Two specific amino acid sequences are thought to represent different binding sites for TSH and for the TSH-R–stimulating antibody (TSH-R [stim] Ab) found in Graves disease (see later).
The amount of thyroid hormone needed to maintain normal organ system function in thyroidectomized individuals is defined as the amount necessary to maintain the plasma TSH within the normal range (0.4–4.8 mU/L). About 80% of orally administered levothyroxine is absorbed from the GI tract, and 100–125 μg/d usually maintains a normal plasma TSH in individuals of average size.
Thyroid hormones exert their actions by two mechanisms: (1) genomic actions mediated by T3 interactions with its nuclear receptors, regulating gene activity; and (2) nongenomic actions effected by T3 and T4 interactions with specific enzymes (such as pyruvate kinase, adenylate cyclase, and calcium APTase), mitochondrial proteins, and glucose transporters. Thyroid hormones enter target tissue cells by either passive diffusion or specific transport carriers through the cell membrane and cytoplasm. Within the cell cytoplasm, most of the T4 is converted to T3. The nuclear receptor for T3 has been cloned and found to be similar to the nuclear receptors for glucocorticoids, mineralocorticoids, estrogens, progestins, vitamin D3, and retinoic acid. For reasons that are unclear, there are two different receptor (TR) genes in humans. Each gene (hTR-α and hTR-β) yields at least two differently spliced proteins: hTR-α (hTR-α1 and hTR α2) and hTR-β (hTR-β1 and hTR-β1). hTR-α2 may be biologically inactive. The TR gene for the alpha form is on chromosome 17 and for the beta form, on chromosome 3. The two different receptor forms may help to explain both the normal variation in thyroid hormone responsiveness of various organs and the selective tissue abnormalities found in various thyroid resistance syndromes. For example, the brain contains mostly α receptors, the liver contains mostly β receptors, and the heart contains both. Point mutations in the hTR-β1 gene result in abnormal T3 receptors and the syndrome of generalized resistance to thyroid hormone (Refetoff syndrome).
When the T3 receptor complex binds to DNA, it increases expression of specific genes, with the induction of related messenger RNAs. A wide variety of enzymes must be produced to account for the many effects of thyroid hormones on cell function.
The effects of thyroid hormones in various organs are summarized in Table 20–1. Thyroid hormones increase the activity of membrane-bound Na+-K+ ATPase, increase heat production, and stimulate oxygen consumption (calorigenesis). Thyroid hormones also affect tissue growth and maturation, help regulate lipid metabolism, increase cardiac contractility by stimulating the expression of myosin protein, and increase intestinal absorption of carbohydrates.
| Target Tissue | Effect | Mechanism |
|---|---|---|
| Heart | Chronotropic | Increase number and affinity of β-adrenergic receptors |
| Inotropic | Enhance responses to circulating catecholamines | |
| Increase proportion of alpha-myosin heavy chain (with higher ATPase activity) | ||
| Lung | Metabolic | Maintenance of ventilator responses to hypoxia and hypercapnia |
| Adipose tissue | Catabolic | Stimulate lipolysis |
| Muscle | Catabolic | Increase protein breakdown |
| Bone | Developmental and metabolic | Promote normal growth and skeletal development; accelerate bone turnover |
| Nervous system | Developmental | Promote normal brain development |
| Gut | Metabolic | Increase rate of carbohydrate absorption, increased gut motility |
| Lipoprotein | Metabolic | Stimulate formation of hepatic LDL receptors |
| Endocrine | Metabolic | Alterations in production, responsiveness and metabolic clearance |
| Other | Calorigenic | Stimulate oxygen consumption by metabolically active tissues (exceptions: adult brain, testes, uterus, lymph nodes, spleen, anterior pituitary) |
| Increase metabolic rate |
The effects of T4 and T3 and the catecholamines epinephrine and norepinephrine are closely interrelated. Both increase the metabolic rate and stimulate the nervous system and heart. In humans, the transcriptional effects of T3 include production of increased numbers of (and perhaps sensitivity to) β-adrenergic receptors in the heart, skeletal muscle, adipose tissue, and lymphocytes.
Checkpoint
Describe a thyroid follicle and its change with activity versus inactivity of the gland.
What forms of thyroid hormone does the thyroid gland secrete? What are the normal proportions of the different forms? What are the relative potencies of each hormone?
To what is thyroid hormone bound during its transport in plasma?
How are thyroid hormone levels regulated?
What is the mechanism of action of thyroid hormone?
What are the most prominent organ system–specific effects of thyroid hormone?
Overview of Thyroid Disease
The symptoms and signs of thyroid disease in humans are predictable consequences of the physiologic effects of thyroid hormones discussed previously. The clinician commonly encounters patients with one of five types of thyroid dysfunction: (1) hyperthyroidism (thyrotoxicosis), caused by an excess of thyroid hormones; (2) hypothyroidism (myxedema), caused by a deficiency of thyroid hormones; (3) goiter, a diffuse enlargement of the thyroid gland, caused by prolonged elevation of TSH; (4) thyroid nodule, a focal enlargement of a portion of the gland, caused by a benign or malignant neoplasm; and (5) abnormal thyroid function tests in a clinically euthyroid patient.
Several laboratory tests are useful in the initial evaluation of patients suspected of having thyroid dysfunction. The first is plasma TSH measured by a sensitive assay (usually defined by a lower detection limit of 0.1 mU/L or less). TSH is below normal in hyperthyroidism and above normal in hypothyroidism (except in the rare instances of pituitary or hypothalamic disease). The second useful laboratory test is measurement of non-protein-bound thyroxine. Most clinical laboratories are now able to accurately measure free thyroxine (FT4) directly. Although rarely used today, an estimate of non-protein-bound thyroxine is provided by the free thyroxine index (FT4I), the product of the total plasma thyroxine (TT4) and the T4 resin uptake (RT4U) (ie, FT4I = TT4 × RT4U). The TT4 by itself often reflects the functional state of the thyroid hormone–binding proteins. The RT4U is an indicator of thyroid-binding globulin and serves to correct for alterations in the concentration of binding protein. Some laboratories instead measure T3 resin uptake (RT3U).
Although total and free T3 levels can be measured, they have a short half-life and are technically difficult assays. Under most circumstances, circulating levels of T3 correlate less well with clinical hyperthyroidism or hypothyroidism.
A variety of thyroid autoantibodies are detectable in patients with thyroid dysfunction, including (1) thyroidal peroxidase antibody (TPO Ab), formerly termed antimicrosomal antibody; (2) thyroglobulin antibody (Tg Ab); and (3) TSH receptor antibody, either stimulating (TSH-R [stim] Ab) or blocking (TSH-R [block] Ab). Thyroglobulin and thyroidal peroxidase antibodies are commonly found in hypothyroidism resulting from Hashimoto thyroiditis and occasionally in hyperthyroidism from Graves disease (see later). TSH-R [stim] Ab is present in individuals with hyperthyroidism caused by Graves disease. Detection of TSH-R [block] Ab in maternal serum is predictive of congenital hypothyroidism in newborns of mothers with autoimmune thyroid disease.
Other procedures such as thyroid scans and the thyrotropin-releasing hormone (TRH) test are discussed later.
Pathophysiology of Selected Thyroid Diseases
The pathogenesis of the most common thyroid diseases probably involves an autoimmune process with sensitization of the host’s own lymphocytes to various thyroidal antigens. Three major thyroidal antigens have been documented: thyroglobulin (Tg), thyroidal peroxidase (TPO), and the TSH receptor. Both environmental factors (eg, viral or bacterial infection or high iodine intake) and genetic factors (eg, defect in suppressor T lymphocytes) may be responsible for initiating autoimmune thyroid disease.
The causes of hyperthyroidism are listed in Table 20–2. Most commonly, thyroid hormone overproduction is due to Graves disease. In Graves disease, the TSH receptor autoantibody TSH-R [stim] Ab stimulates the thyroid follicular cells to produce excessive amounts of T4 and T3. Less commonly, patients with multinodular goiter may become thyrotoxic without circulating antibodies if given inorganic iodine (eg, potassium iodide) or organic iodine compounds (eg, the antiarrhythmic drug amiodarone, which contains 37% iodine by weight). Multinodular goiters may also develop one or more nodules that become autonomous from TSH regulation and secrete excessive quantities of T4 or T3. Patients from regions where goiter is endemic may develop thyrotoxicosis when given iodine supplementation (jodbasedow phenomenon). Large follicular adenomas (>3 cm in diameter) may produce excessive thyroid hormone.
| Etiologic Classification | Pathogenetic Mechanism |
|---|---|
| Thyroid hormone overproduction | |
| Graves disease | Thyroid-stimulating hormone receptor–stimulating antibody (TSH-R [stim] Ab) |
| Toxic multinodular goiter | Autonomous hyperfunction |
| Follicular adenoma | Autonomous hyperfunction |
| Pituitary adenoma | TSH hypersecretion (rare) |
| Pituitary insensitivity | Resistance to thyroid hormone (rare) |
| Hypothalamic disease | Excess TRH production |
| Germ cell tumors: choriocarcinoma, hydatidiform mole | Human chorionic gonadotropin stimulation |
| Struma ovarii (ovarian teratoma) | Functioning thyroid elements |
| Metastatic follicular thyroid carcinoma | Functioning metastases |
| Thyroid gland destruction | |
| Lymphocytic thyroiditis | Release of stored hormone |
| Granulomatous (subacute) thyroiditis | Release of stored hormone |
| Hashimoto thyroiditis | Transient release of stored hormone |
| Drug effect | |
| Thyrotoxicosis medicamentosa, thyrotoxicosis factitia | Ingestion of excessive exogenous thyroid hormone |
| Amiodarone | Excess iodine and/or thyroiditis |
| Interferon alpha | Thyroiditis |
Occasionally, TSH overproduction (eg, from a pituitary adenoma) or hypothalamic disease may cause excessive thyroid hormone production. The diagnosis is suggested by clinically evident hyperthyroidism with elevated serum T4 and T3 and elevated serum TSH levels. Neuroradiologic procedures such as computed tomography (CT) scans or magnetic resonance imaging (MRI) of the sella turcica confirm the presence of a pituitary tumor. Even more rarely, hyperthyroidism results from TSH overproduction caused by pituitary (but not peripheral tissue) resistance to the suppressive effects of T4 and T3. The diagnosis is suggested by finding elevated serum T4 and T3 levels with an inappropriately normal serum TSH level.
Hyperthyroidism may be precipitated by germ cell tumors (choriocarcinoma and hydatidiform mole), which secrete large quantities of human chorionic gonadotropin (hCG). The large quantities of hCG secreted by these tumors bind to the follicular cell TSH receptor and stimulate overproduction of thyroid hormone. Rarely, hyperthyroidism can be produced by ovarian teratomas containing thyroid tissue (struma ovarii). Hyperthyroidism results when this ectopic thyroid tissue begins to function autonomously. Patients with large metastases from follicular thyroid carcinomas may produce excess thyroid hormone, particularly after iodide administration.
Transient hyperthyroidism is occasionally observed in patients with lymphocytic or granulomatous (subacute) thyroiditis (Hashimoto thyroiditis). In such cases, the hyperthyroidism is due to destruction of the thyroid with release of stored hormone.
Finally, patients who consume excessive amounts of exogenous thyroid hormone (accidentally or deliberately) and those treated with amiodarone or interferon alpha may present with symptoms, signs, and laboratory findings of hyperthyroidism.
Whatever the cause of hyperthyroidism, serum thyroid hormones are elevated. Both the free thyroxine (FT4) and the free thyroxine index (FT4I) are elevated. In 5–10% of patients, T4 secretion is normal while T3 levels are high (so-called T3 toxicosis). Total serum T4 and T3 levels are not always definitive because of variations in concentrations of thyroid hormone–binding proteins.
Hyperthyroidism resulting from Graves disease is characterized by a suppressed serum TSH level as determined by sensitive immunoenzymometric or immunoradiometric assays. However, TSH levels may also be suppressed in some acute psychiatric and other nonthyroidal illnesses. In the rare TSH-secreting pituitary adenomas (so-called secondary hyperthyroidism) and in hypothalamic disease with excessive TRH production (so-called tertiary hyperthyroidism), hyperthyroidism is accompanied by elevated plasma TSH.
The radioactive iodine (RAI) uptake of the thyroid gland at 4, 6, or 24 hours is increased when the gland produces an excess of hormone (eg, Graves disease); it is decreased when the gland is leaking stored hormone (eg, thyroiditis), when hormone is produced elsewhere (eg, struma ovarii), and when excessive exogenous thyroid hormone is being ingested (eg, factitious hyperthyroidism). Technetium 99m scanning can provide information similar to that obtained with RAI and is quicker and entails less radiation exposure.
The TRH test is sometimes helpful in diagnosis when patients have confusing results of thyroid function tests. In normal individuals, administration of TRH (500 μg intravenously) produces an increase in serum TSH of at least 6 mU/L within 15–30 minutes. In primary hyperthyroidism, TSH levels are low and TRH administration induces little or no rise in the TSH level.
Graves disease is the most common cause of hyperthyroidism. In this condition, the thyroid gland is symmetrically enlarged and its vascularity markedly increased. The gland may double or triple in weight. Microscopically, the follicular epithelial cells are columnar in appearance and increased in number and size (Figure 20–2). The follicles are small and closely packed together. The colloid is scanty; the edges are scalloped in appearance secondary to the rapid proteolysis of thyroglobulin. The gland’s interstitium is diffusely infiltrated with lymphocytes and may contain lymphoid follicles with germinal centers.
The serum of more than 90% of patients with Graves disease contains TSH-R [stim] antibody, directed against the TSH receptor site in the thyroid follicular epithelial membrane. This antibody, formerly called long-acting thyroid stimulator (LATS), is now also called thyroid-stimulating immunoglobulin (TSI). When it binds to the cell membrane TSH receptors, TSH-R [stim] Ab stimulates hormone synthesis and secretion in somewhat the same way as TSH. Although serum levels of TSH-R [stim] Ab correlate poorly with disease severity, its presence can be helpful diagnostically and perhaps prognostically. After discontinuation of antithyroid drug treatment, about 30–50% of patients with Graves hyperthyroidism relapse. There seems to be a greatly increased recurrence risk if the TSH-R [stim] Ab is still found in plasma at the time of discontinuation of the antithyroid drug treatment, so this test can perhaps be used to predict likely relapse.
The genesis of TSH-R [stim] Ab in patients with Graves disease is uncertain. However, Graves disease is familial. A genetic contribution to the development of Graves disease is suggested by the finding of much higher concordance rates in monozygotic same-sex twin pairs (0.35) than in dizygotic pairs (0.03). In Caucasians, it is associated with the HLA-B8 and HLA-DR3 histocompatibility antigens; in Asians, with HLA-Bw46 and HLA-B5; and in blacks, with HLA-B17. Furthermore, patients with Graves disease frequently suffer from other autoimmune disorders (Table 20–3). The precipitating cause of this antibody production is unknown, but an immune response against a viral antigen that shares homology with TSH receptor may be responsible. Another theory of the pathogenesis of Graves disease is a defect of suppressor T lymphocytes, which allows helper T lymphocytes to stimulate B lymphocytes to secrete antibodies directed against follicular cell membrane antigens, including the TSH receptor (Figure 20–6).
|
|
|
|
Figure 20–6
Proposed pathogenesis of Graves disease. A defect in suppressor T lymphocytes (Ts) allows helper T lymphocytes (TH) to stimulate B lymphocytes (B) to synthesize thyroid autoantibodies. The thyroid receptor–stimulating antibody (TSH-R [stim] Ab) is the driving force for thyrotoxicosis. Inflammation of the orbital muscles may be due to sensitization of cytotoxic T lymphocytes (Tc), or killer cells, to orbital antigens linked to an antigen in the thyroid. What triggers this immunologic cascade is not known. Ag, antigen; P Ab, peroxidase or microsomal antibody; Tg Ab, thyroglobulin antibody. (Redrawn, with permission, from Gardner DG et al, eds. Greenspan’s Basic and Clinical Endocrinology, 9th ed. McGraw-Hill, 2011.)
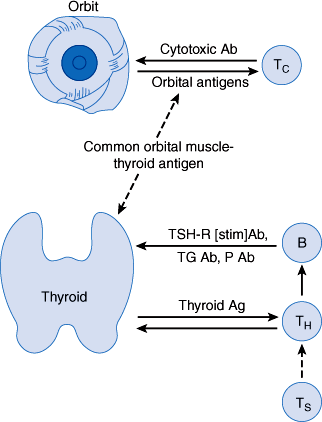
Moderate titers of other autoantibodies (thyroidal peroxidase antibody and TSH-R [block] Ab) can be found in patients with Graves disease. Their significance is uncertain. In some cases, TSH-R [block] Ab appears after 131I radioiodine therapy of Graves disease.
Patients with hyperthyroidism from Graves disease may later develop hypothyroidism by one of several mechanisms: (1) thyroid ablation by surgery or 131I radiation treatment; (2) autoimmune thyroiditis, leading to thyroid destruction; and (3) development of antibodies that block TSH stimulation (TSH-R [block] Ab).
After radioactive iodine therapy, there is often a lag in recovery of thyrotropin (TSH) responsiveness that may last 60–90 days or longer. During this period, decisions regarding further therapy must be based on the patient’s clinical status as well as on the serum levels of TSH and thyroid hormones.
Checkpoint
What are the five categories of thyroid dysfunction most commonly observed in patients?
What are seven different pathophysiologic mechanisms by which a patient might develop hyperthyroidism?
What is the most useful initial test of thyroid function in hyperthyroidism? What results would you expect compared with normal?
How can thyroid scanning help confirm the suspected cause of hyperthyroidism?
Describe the mechanism of hyperthyroidism in Graves disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


