INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should understand:
The physiology of the natural anticoagulant mechanisms and how they limit the generation of activated enzymes in the coagulation cascade.
How genetic defects in natural anticoagulant mechanisms and abnormalities in procoagulant factors can predispose to venous thrombosis.
The interaction of acquired and genetic risk factors in the pathogenesis of venous thrombosis.
As mentioned in the introduction to this section of the book, thrombosis plays a commanding role as a complication of highly prevalent disorders such as obesity, diabetes, and cancer. Myocardial infarction is the leading cause of mortality from arterial thrombosis, whereas pulmonary embolus accounts for most deaths due to venous thrombosis. In this chapter, we will stress the pathophysiologic principles underlying in vivo thrombus formation and the acquired and inherited factors that put patients at increased risk.
Insight into the pathogenesis of thrombotic disorders began with the 19th century pathologist Rudolf Virchow, whose clinical and postmortem observations led him to postulate that the formation of blot clots sufficiently large to occlude major blood vessels was dependent upon three independent processes:
Abnormal blood flow—principally stasis.
Irritation of the vessel wall due to trauma, or a degenerative/inflammatory process such as atherosclerosis.
Enhanced blood coagulation, or what we now call hypercoagulability.
Virchow’s triad provides a useful framework for evaluating any patient who has developed clinically significant thrombotic disease. The history will often reveal factors that predispose to stasis, such as immobilization of a limb, pregnancy, or prolonged inactivity. Further studies may identify a specific type of vascular pathology as well as abnormalities in blood coagulation. In this chapter, venous thrombosis will be covered in more depth than arterial thrombosis because the former is closely linked to well-defined abnormalities in coagulation, whereas the latter is much more dependent on vascular abnormalities, particularly atherosclerosis, which lie outside the scope of this book.
As outlined in Table 17-1, both nature and nurture contribute to the development of venous thrombosis. As listed in the left hand column, a wide spectrum of acquired or secondary factors puts individuals at increased risk. Several, such as increasing age, male sex, and obesity, are weak but statistically significant risk factors. In contrast, a history of venous thrombosis is a strong predictor of repeated episodes. Immobilization due to extended bed rest or prolonged air travel leads to blood stasis, particularly in the lower extremities. Immobilization is also an important contributor to the increased risk of venous thrombosis in patients recovering from major surgery. Pregnancy causes stasis in the legs, owing to impairment of venous return by the enlarging uterus. Malignancy and estrogen use will be discussed later in the chapter along with other less common acquired risk factors.
| Acquired (Secondary) | Inherited (Primary) |
|---|---|
Advancing Age Male Sex Obesity Prior Thrombosis Immobilization Pregnancy Major Surgery Malignancy Estrogens (OCP* etc) Antiphospholipid Antibody Syndrome Heparin-Induced Thrombocytopenia Myeloproliferative Disorders Inflammatory Bowel Disease | Factor V Leiden (FVL) Prothrombin G20210A Antithrombin Deficiency Protein C Deficiency Protein S Deficiency |
INHERITED DEFECTS THAT ENHANCE THE RISK OF VENOUS THROMBOSIS
During the last 15 years, it has become apparent that inherited abnormalities of blood coagulation play an important role in determining which individuals are at a higher risk for development of venous thrombosis. The right-hand column of Table 17-1 lists monogenic defects linked to the development of venous thrombosis. All consist of mutations in clotting factors or in inhibitors of clotting. Figure 17-1 depicts where these proteins are situated in the coagulation cascade.
FIGURE 17-1
Proteins that participate in the control or limitation of clot formation. Familial thrombophilia can be caused by mutations that affect antithrombin, prothrombin (Pro), factor V, protein C, and protein S. The “a” signifies activation; thus Ca = activated protein C. The circle with the X indicates inhibition or inactivation. EPCR: endothelial protein C receptor.
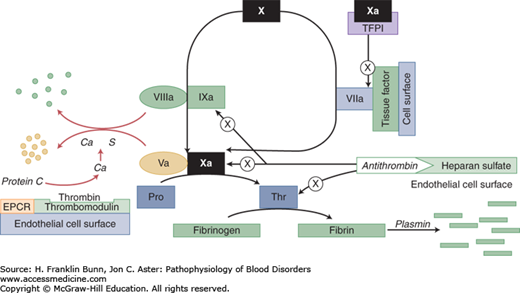
Two of these genetic variants are mutations in genes encoding clotting proteins—factor V and prothrombin:
Factor V Leiden is a genetic variant created by a single base change that results in the replacement of arginine by glutamine at position 506. In normal factor Va, Arg506 is the first site cleaved by activated protein C (Fig. 17-2). Subsequent cleavage by this serine protease at Arg306 and Arg679 inactivates factor Va. The absence of the initial cleavage at position 506 in factor V Leiden markedly delays cleavage at the two other sites and thereby results in higher levels of factor Va, which acts as a cofactor for factor Xa.
Prothrombin G20210A is a single base substitution in the 3′ untranslated region of the prothrombin gene that increases the biosynthesis of prothrombin protein. As a result, individuals heterozygous for this mutation have approximately 30% higher plasma levels of structurally normal prothrombin compared with normal individuals. This seemingly modest increase in concentration significantly increases the risk of venous thrombosis.
FIGURE 17-2
Proteolytic degradation of the heavy chain of factor Va by activated protein C. (APC) Normal factor Va is rapidly cleaved at Arg506 with subsequent cleavage at Arg306 and Arg679. In the factor V Leiden mutation, the absence of the Arg cleavage site at 506 greatly delays subsequent cleavage, K: rate of cleavage.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


