INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should have a coherent understanding of:
The organization and function of the major proteins of the red cell membrane and cytoskeleton.
The pathogenesis and clinical features of hereditary spherocytosis.
The genetics and pathogenesis of glucose-6-phosphate dehydrogenase deficiency.
DISORDERS OF THE RED CELL MEMBRANE
As mentioned in Chapter 1, the red cell has two relatively simple yet highly important responsibilities—the transport of oxygen from the lungs to respiring organs and tissues and the transport of carbon dioxide in the reverse direction. Because of its relatively long life span of 120 days, a single red cell makes about 170,000 circuits through the microcirculation and, in doing so, travels roughly 100 miles! The efficiency of both gas transport and flow through narrow channels in the capillary and splenic circulation is enhanced by the ability of the red cell to change shape. Thus, in order for a red cell to fulfill its functions, its membrane must have a high degree of durability and flexibility.
The basis for the remarkable mechanical properties of the red cell is a network of proteins that assemble to form a specialized membrane skeleton, depicted in Figure 10-1. The primary building blocks of the membrane skeleton are alpha (α)-spectrin and beta (β)-spectrin, which bind to each other to form long, stable dimers. Self-association of spectrin dimers and docking of spectrin to a complex of actin filaments, with the aid of protein 4.1, creates a flexible, branching, hexagonal network that covers the entire inner surface of the membrane. The membrane skeleton is tethered to the membrane’s lipid bilayer by vertical interactions with a complex composed of the anion exchange channel (band 3), ankyrin, and protein 4.2. These horizontal and vertical interactions within the membrane skeleton and with the lipid bilayer are critical determinants of the pliability and tensile strength of the red cell membrane.
FIGURE 10-1
Cross-section of the red cell membrane. The membrane skeleton is composed principally of spectrin (green), which binds to itself at one end and attaches to short filaments of F-actin (blue) at the other end, aided by protein 4.1 (orange). Up to six spectrins can bind to one actin filament, making the skeleton a hexagonal array. The membrane skeleton is attached to band 3 (pink), the anion exchanger, via ankyrin near the spectrin self-association site, and to band 3 via proteins 4.1 and 4.2 (yellow) at the actin end of spectrin. Other proteins (gray) participate in large protein complexes at each of these sites. Defects in the “vertical” connections between the membrane skeleton and band 3 result in hereditary spherocytosis. Defects in the “horizontal” interactions that hold the membrane skeleton together cause hereditary elliptocytosis or its severe variant, hereditary pyropoikilocytosis.(Modified with permission from Grace RF and Lux SE, Disorders of the Red cell membrane. In: Orkin SH, Nathan DG, eds. Nathan and Oski’s Hematology of Infancy and Childhood. 5th ed. Philadelphia, USA, Saunders; 2008: 671.)
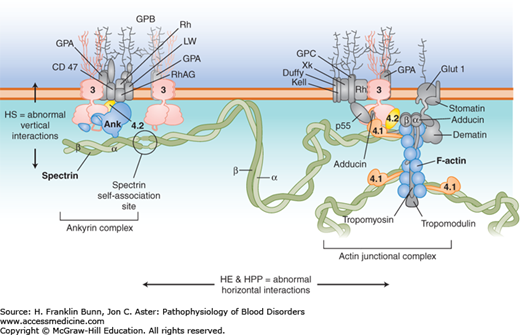
As discussed in the following sections, molecular defects in vertical interactions cause hereditary spherocytosis, whereas defects in horizontal interactions result in the formation of elliptical red cells.
Under the microscope, normal red cells appear as biconcave discs. Adoption of this shape requires a significant amount of excess membrane surface area and gives the red cell considerable deformability, allowing it to traverse narrow strictures in the microcirculation, especially within the spleen. In contrast, a cell with a minimal ratio of surface area to volume would assume the shape of a sphere and be non-deformable. A relatively simple laboratory test called osmotic fragility can provide an accurate assessment of the surface area to volume ratio of red cells (Fig. 10-2). When exposed to solutions of decreasing salt (NaCl) concentration, the red cell swells progressively until it becomes a sphere. With further reduction in salt concentration, the non-distensible membrane ruptures, leading to release of the cell’s content of hemoglobin. Spherocytes have a low ratio of surface area to volume and undergo lysis at a higher salt concentration than that required for lysis of normal cells. In contrast, target cells, which have a higher surface area to volume ratio, undergo lysis at a lower salt concentration.
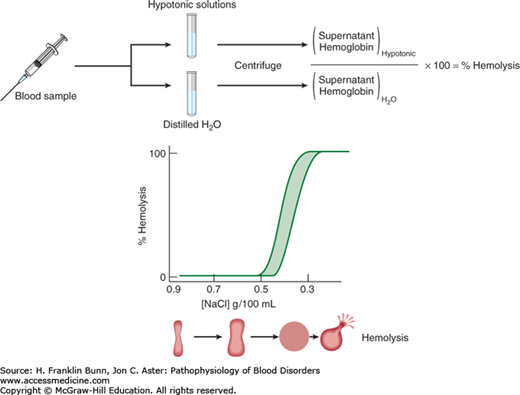
In patients with hereditary spherocytosis (HS), the presence of non-deformable spherocytes causes hemolysis of varying severity. It is one of the most commonly encountered inherited hemolytic anemias, with an incidence of about 1 in 5000. In the great majority of cases, an autosomal dominant inheritance pattern is apparent. In most of these patients, the causative mutations involve ankyrin, band 3, or β-spectrin. Those with recessive HS owing to biallelic mutations in α-spectrin have much more severe hemolysis. In contrast, individuals with biallelic mutations in protein 4.2 have mild HS. As shown in Figure 10-1, all of these proteins participate in the “vertical” assembly of a complex that stabilizes the interaction of the membrane skeleton with the lipid bilayer. Mutations
that result in either impaired production or, less commonly, altered function of one of these proteins cause shedding of membrane microvesicles and thus a decrease in the ratio of surface area to volume. These rigid spherocytes are trapped in the spleen, where they are either destroyed or “conditioned” to become even more spherocytic.
The severity of HS varies considerably. Although all affected individuals have hemolysis with increased reticulocyte counts, those with mild anemia (compensated hemolysis) have minimal or no symptoms and often are not diagnosed until later in life. Most patients have a modestly enlarged, palpable spleen. Those with more marked hemolysis have symptoms of severe anemia and are often mildly icteric. Like other patients with chronic hemolysis, patients with HS, even adolescents or young adults, commonly develop gallstones caused by the precipitation of poorly soluble nonconjugated bilirubin, serum levels of which are elevated owing to the enhanced rate of red cell destruction, as explained in Chapter 3. Patients with HS may have sudden worsening of anemia because of either acute suppression of erythropoiesis, such as during parvovirus B19 infection, or enhanced hemolysis, such as during the transient splenic hyperplasia that accompanies infectious mononucleosis.
The diagnosis of HS should be suspected in any patient with life-long hemolytic anemia, especially when family members are also anemic. In moderate or severe cases, spherocytes are readily detected in the peripheral blood film, appearing as small, dark, red cells with no central pallor (Fig. 10-3). When the hemolysis is mild, the spherocytes are less apparent and are often overlooked. Consistent with the dark red color of spherocytes seen on the blood film, the mean cell hemoglobin concentration (MCHC) is increased in a portion of the red cells. Spherocytes are also the prime morphologic feature of an acquired disorder, immune hemolytic anemia (Chapter 11).The diagnosis of HS can be confirmed by the osmotic fragility test, as described in Figure 10-2. The presence of osmotically fragile cells, that is, those with a decreased surface area to volume ratio, can be enhanced by a 24-hour incubation period at 37°C before the test is performed.
FIGURE 10-3
Blood film: hereditary spherocytosis.



