INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should be able to:
Distinguish among the different causes of thrombocytopenia.
Understand the pathogenesis of immune thrombocytopenic purpura and the principles of treatment.
Understand the pathogenesis of thrombotic thrombocytopenic purpura and the principles of treatment.
Define mechanisms that lead to qualitative platelet defects.
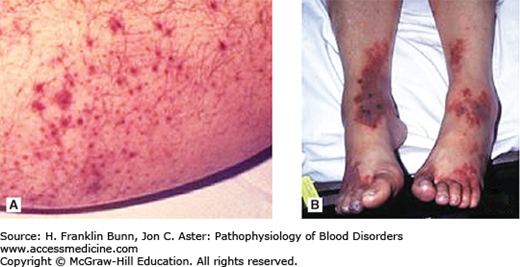
Platelets assume an important role in a wide variety of disease states. Morbidity related to platelet dysfunction is generally due to bleeding, but occasionally thrombosis is dominant. As summarized in Table 14-1, bleeding can occur if the platelet count is too low or if there is a qualitative defect in platelet function. In patients with platelet disorders, bleeding is usually superficial and localized to skin and mucous membranes, a phenomenon known as purpura. Petechiae are small, 2-to-5 mm, red or purple macules (spots) that appear most often in the distal lower extremities (Fig. 14-1A) but also on the conjunctiva and palate. Small petechiae often merge to form larger lesions. Very large, purpuric lesions are called ecchymoses (Fig. 14-1B). In contrast to telangiectasia, petechiae and ecchymoses do not blanch under pressure. Mucosal purpura is generally associated with severe thrombocytopenia and may be a harbinger of complications such as gastrointestinal bleeding or even brain hemorrhage. In contrast to the superficial bleeding seen in patients with thrombocytopenia and qualitative platelet disorders, defects in the soluble coagulation factors, such as hemophilia (Table 14-1), typically present with deep bleeding, such as hemarthroses.
Platelet defects:
Superficial bleeding
Petechiae, ecchymoses
Coagulation factor defects:
Deep bleeding
Hemarthrosis, etc
| Disorder | Etiology | Occurrence | Examples |
|---|---|---|---|
| Bleeding | |||
| Thrombocytopenia | Acquired | Common | ITP, DIC, marrow aplasia or malignancy |
| Qualitative defect | Acquired | Common | Drugs, uremia, bone marrow disorders |
| Hereditary | Rare | Bernard-Soulier syndrome, Glanzmann thrombasthenia | |
| Thrombosis | |||
| Thrombocytosis | Acquired | Uncommon | Myeloproliferative disorders |
THROMBOCYTOPENIA
In a clear and informative parallel to anemia, thrombocytopenia can be due either to decreased platelet production or enhanced platelet destruction. The life span of normal platelets in the circulation is about 7 to 9 days. Therefore, if the bone marrow stops producing platelets, it takes nearly a week before severe thrombocytopenia develops. In contrast, an acute immune or consumptive process that suddenly and drastically curtails platelet survival can produce severe thrombocytopenia within hours.
Damage or suppression of pluripotent hematopoietic stem cells may result in not only thrombocytopenia but also anemia and leukopenia (pancytopenia) accompanied by marrow aplasia. Aplastic anemia and other causes of pancytopenia are covered in Chapter 4. Transient thrombocytopenia due to marrow aplasia and decreased platelet production predictably follows certain forms of chemotherapy or radiation therapy for cancer. Other drugs selectively inhibit platelet production. Occasionally, patients with severe alcoholism will present with thrombocytopenia that appears to be due to a toxic effect of ethanol on megakaryocytes. In most cases, the platelets gradually return to baseline levels following cessation of therapy or drinking. Somewhat less often, decreased platelet production is due to a primary bone marrow disorder such as acute leukemia or a myelodysplastic syndrome. Less severe thrombocytopenia can be due to megaloblastic anemia or to invasion of the bone marrow by tumors such as lymphoma or carcinoma.
The administration of thrombopoietin (Tpo) or Tpo-mimetic agents is seldom effective in the treatment of severe thrombocytopenia due to impaired platelet production. This may be due to the fact that endogenous plasma Tpo levels are already markedly elevated in these patients, owing to very low numbers of megakaryocytes in the bone marrow as well as platelets in the periphery. (See Chapter 2 for information on the regulation of platelet production by endogenous Tpo.) Because the platelet life span is usually normal in these patients, platelet transfusions produce a significant and beneficial rise in the platelet count unless or until the patient develops alloimmunization. In contrast, platelet transfusions are much less effective in patients in whom thrombocytopenia is due to enhanced destruction.
As explained in Chapter 1, the spleen in normal individuals contains (sequesters) about a third of the body’s platelets. Patients who have splenomegaly due to a wide range of disorders (Chapter 1, Table 1-4) have a higher proportion of their platelets entrapped along with white and red blood cells. A significant reduction in one or more of the peripheral blood counts by splenic sequestration is called hypersplenism.
Patients with hypersplenism generally have platelet counts of 50,000 to 120,000/mm3. Thus, the degree of thrombocytopenia is almost never low enough to pose a risk of hemorrhage. Accordingly, splenectomy is not indicated for correction of the platelet count. However, some patients may have neutropenia that is sufficiently severe to warrant splenectomy.
Severe thrombocytopenia is commonly caused by rapid destruction of platelets, most often due either to immune clearance or to consumption in conditions associated with intravascular thrombosis. In either case, the bone marrow produces increased numbers of platelets that have a short survival in the circulation. Because platelets normally become smaller as they age in vivo, the few platelets that are seen in blood films of patients with thrombocytopenia due to increased destruction tend to be larger than average.
Immunologic recognition followed by destruction of platelets is a relatively common cause of severe thrombocytopenia in individuals of all age groups. Most of these individuals come to medical attention because they develop purpura—hence the designation immune thrombocytopenic purpura (ITP). In children, the peak incidence is about 5 years of age. These previously healthy children typically develop a sudden onset of petechiae or ecchymoses within a few days or weeks following an infectious illness, usually viral. In the majority of these patients, the thrombocytopenia and purpura resolve within 6 months regardless of whether they have received therapy. Adults with ITP have a strikingly different clinical presentation and course. In these patients, the onset is insidious and seldom accompanied by a viral prodrome. The illness is chronic and, when severe, requires meticulous and often complex management. In both children and adults, if ITP is the primary diagnosis, patients normally have no symptoms or physical findings except for purpura. The spleen and lymph nodes are not enlarged. Some women also experience menorrhagia. Gastrointestinal bleeding is much less common. Rarely patients with ITP sustain cerebral hemorrhage.
There is overwhelming circumstantial evidence that in ITP platelets are destroyed by autoantibodies directed against platelet antigens. In early (less regulated) days of clinical research, it was noted that the intravenous administration of plasma from a patient with ITP to a normal volunteer resulted in the development of acute and severe thrombocytopenia and purpura. These studies led to the hypothesis that ITP patients had circulating autoantibodies directed against “public” antigens displayed on the surface of platelets in all individuals. These antibody-coated platelets are rapidly cleared by the interaction of the Fcγ receptor on tissue macrophages with the Fc (constant) portion of the immunoglobulin.
The primary public antigen targeted for immune attack in ITP is GPIIb/IIIa, which, as explained in Chapter 13, undergoes a conformational change during platelet activation and is the site of fibrinogen cross-linking. As shown in Figure 14-2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


