Thoracoscopic Repair of Esophageal Atresia and Tracheoesophageal Fistula
George W. Holcomb, III
The earliest description of a baby with esophageal atresia (EA) has been attributed to William Durston in 1670. However, it was not until 1939 that a baby survived following operative intervention. On successive days in November 1939, two different babies in two different cities underwent an initial gastrostomy followed, at a later date, by esophageal substitution. The first successful primary repair was performed by Dr. Cameron Haight in Ann Arbor in 1941 through a left thoracotomy. The baby developed an anastomotic leak and stricture, but required only one esophageal dilatation. She stayed in the hospital for many months, but recovered uneventfully and led a normal life. Interestingly, she had a son who was born with EA and a distal fistula.
EA is really a spectrum of anomalies. A number of classification systems (Gross, Vogt, etc.) have been described. However, it seems best to describe the anomaly rather than classify it (Fig. 1). EA with an associated tracheoesophageal fistula (TEF) is the most common variant of the EA spectrum of diseases and occurs in about 85% to 88% of all EA cases. Isolated EA (without an associated TEF) occurs in about 7% to 8% of the cases. EA with a proximal fistula is found in 1% to 2% of the cases. The fistula is similar to the H-type TEF and starts proximally on the trachea and ends distally in the dilated proximal esophagus. EA with both a proximal and distal TEF is found in 1% of the cases or less. Although not technically part of the EA classification, an H-type TEF is considered part of the spectrum because these patients also have one or more associated anomalies related to the VACTERL syndrome (Vertebral, Atresias (GI tract), Cardiac, Tracheoesophageal (TE), Renal, Limb). This isolated TEF is usually located in the lower cervical region or upper thoracic cavity and is almost always amenable to repair through a cervical approach. The H-type TEF is actually the third most common of these anomalies.
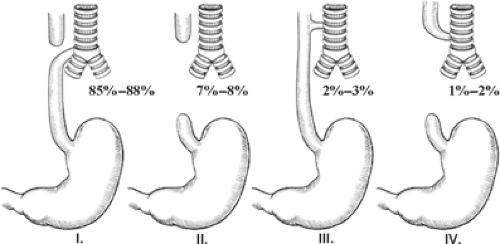 Fig. 1. This schematic depicts the spectrum of anomalies seen in patients with esophageal atresia (EA) and tracheoesophageal fistula (TEF). The incidence of each anomaly is also noted. The most common anomaly is EA with a distal TEF (I). Next in incidence is EA without a TEF (II). Third is an H-type TEF (III). EA with a proximal (but not distal) TEF occurs in about 1% of the cases (IV). |
Embryology
It remains controversial as to the exact etiology for the development of EA/TEF. Around the fourth week of gestation, the foregut begins to differentiate into a dorsal esophageal part and a ventral respiratory segment. Originally, it was felt that the ventral respiratory segment separates from the esophagus by the formation of lateral tracheoesophageal folds which fuse in the midline to create the tracheoesophageal septum. This separation of the trachea and esophagus is usually complete by six to seven weeks of gestation. If there is incomplete fusion of these tracheoesophageal folds, a defective tracheoesophageal septum could develop with an abnormal connection between the trachea and esophagus. However, recently, chick embryo studies have not demonstrated these lateral tracheoesophageal folds. Instead, cranial and caudal folds were found in the region of the tracheoesophageal separation. According to the second theory, EA/TEF could develop due to an imbalance in the growth of these cranial and caudal folds. A third
theory is based on a defect in intracellular signaling due to a deficiency in the sonic hedgehog (Shh) genes. In culture experiments, the fistula tract has been induced to branch by exogenous administration of the Shh signaling molecule and its receptor. These studies support the fistula as arising from the respiratory tract because the normal lung, but not normal esophagus, branches in response to the Shh signaling. Finally, from rat studies, a fourth theory postulates that EA/TEF might result from disturbances in either apoptosis or epithelial proliferation.
theory is based on a defect in intracellular signaling due to a deficiency in the sonic hedgehog (Shh) genes. In culture experiments, the fistula tract has been induced to branch by exogenous administration of the Shh signaling molecule and its receptor. These studies support the fistula as arising from the respiratory tract because the normal lung, but not normal esophagus, branches in response to the Shh signaling. Finally, from rat studies, a fourth theory postulates that EA/TEF might result from disturbances in either apoptosis or epithelial proliferation.
Epidemiology
EA is usually a sporadic occurrence. The incidence of EA/TEF is about 1 per 3,500 births. There appears to be a slight male predominance of 1.3. Among parents who have one affected child, the risk of another affected child is between 0.5% and 2%. In addition, the risk of a child with EA/TEF born to a mother or father with EA/TEF is 3% to 5%. A number of etiologic agents have been identified including medications such as progesterone and estrogen, maternal diabetes, thalidomide exposure, the use of methimazole in early pregnancy, and the prolonged use of contraceptive pills, among others. Also, chromosomal anomalies including Trisomy 18 and 21 have been identified in 5% to 10% of the cases. Finally, defects in three different genes have been found to be associated with EA/TEF.
Associated Anomalies
As previously mentioned, the VACTERL syndrome is the strongest syndromic association with the entire EA spectrum of conditions. The frequencies of these associated conditions are as follows: vertebral (6% to 20%), atresias of the intestinal tract (10% to 15%), cardiac (15% to 35%), renal (5% to 15%), and limb (5% to 20%). EA has also been seen occasionally in patients with the CHARGE syndrome (Coloboma, Heart anomalies, Atresia of the choanae, Retardation, Genital hypoplasia, and Ear deformities).
In a 2007 paper from our institution, we reviewed the incidence of the VACTERL association in 112 patients with EA anomalies seen between 1985 and 2005. There were 62 males and 50 females. In this review, 24.1% of the babies had vertebral anomalies, 14.3% had intestinal atresias, and 32.1% had cardiac anomalies, which was defined as an intrinsic heart lesion other than a patent foramen ovale or patent ductus arteriosus. The most common cardiac condition was a ventriculoseptal defect (VSD) found in 22.3% cases. However, a VSD was found as an insolated condition in only 7.1% of the patients. Cyanotic heart disease was found in only 4.5% of the babies and all were tetralogy of Fallot. Of these 112 babies, renal anomalies occurred in 17% and skeletal in 16.1%. A distal TEF was found in 95.5% of the babies.
Antenatal Diagnosis
On occasion, the diagnosis of EA with or without a fistula can be suspected antenatally. The two findings that can be attributed to EA on prenatal ultrasound (US) are the development of maternal polyhydramnios and an absent or small gastric bubble. However, as polyhydramnios and a small stomach bubble can be found in a number of fetal anomalies, these findings are not specific for EA. Regardless, their association together does raise the possibility of EA in the fetus. Magnetic resonance imaging (MRI) is a new modality for looking at fetal thoracic conditions. In one study looking at 10 fetuses that were thought to be at risk for EA on the basis of prenatal US findings, the MRI was 100% sensitive for the identification of the five EA/TEF babies.
Postnatal Diagnosis
Postnatally, the diagnosis is usually first suspected when the baby has feeding difficulties and a nasogastric tube cannot be passed into the stomach. In babies with known polyhydramnios or other stigmata of the VACTERL syndrome, a nasogastric tube should be passed before feeding, thereby excluding the diagnosis of EA. If the nasogastric tube cannot be passed, a plain radiograph is obtained. With EA, the tip of the catheter is usually visualized in the superior mediastinum at the T2–T4 level (Fig. 2). It is important to identify whether air is seen in the gastrointestinal tract, thereby indicating the presence of a distal TEF, whereas a gasless abdomen would be seen in a baby with isolated EA. Interestingly, there have been scattered reports describing a baby with EA and distal TEF with mucous occluding the distal fistula. In such a baby, the abdomen may be gasless on the plain radiograph.
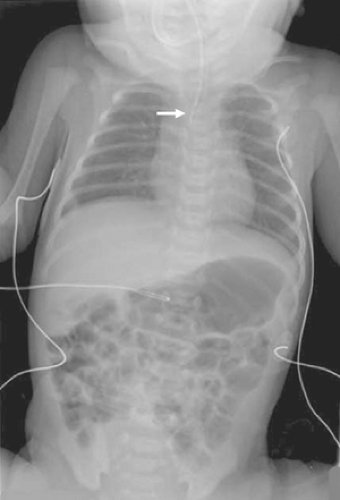 Fig. 2. This baby developed coughing and feeding intolerance shortly after birth. A nasogastric tube was introduced, but could not be advanced past the upper thoracic esophagus (arrow). Air is noted in the stomach, small intestine, and colon. This is a classic radiograph for a baby with EA and a distal TEF. |
Once the diagnosis is made, further workup for VACTERL associations is important. In addition to the physical examination that may identify anorectal or limb anomalies, renal and spinal US are necessary. Also, a cardiac echo or thoracic US should be performed to identify the location of the aortic arch, as the operative approach
ideally should be on the side opposite to the aortic arch. In approximately 5% of the babies, the aortic arch will descend on the right side, making a right thoracic approach much more difficult. In such a patient, a left thoracic approach should be considered.
ideally should be on the side opposite to the aortic arch. In approximately 5% of the babies, the aortic arch will descend on the right side, making a right thoracic approach much more difficult. In such a patient, a left thoracic approach should be considered.
In a term baby, EA or EA/TEF is rarely an emergency and there is usually adequate time to complete the preoperative workup and plan the operative approach. However, in the preterm infant with respiratory distress syndrome requiring mechanical ventilation, the sudden inability to ventilate the baby can be encountered due to the preferential flow of the ventilated gas through the endotracheal tube, down the trachea, and out through the distal TEF. This flow pattern results in poor pulmonary inflation and abdominal distention, both of which inhibit adequate ventilation. Gastric and duodenal perforations have been described. In such cases, a variety of maneuvers have been successful, including immediate gastrostomy with placement of the tube to underwater drainage, immediate thoracotomy and ligation of the fistula, and attempted placement of a Fogarty balloon past the endotracheal tube (ETT) and into the fistula, thereby occluding it. Isolated case reports on the management of babies with this complication can be found in the literature.
While the infant is awaiting operation, it is important to prevent complications resulting from aspiration. The baby should be placed in a head-up position and tilted to one side. Also, it is important that the nasoesophageal catheter is on constant suction. Finally, the baby’s mouth and pharynx should be suctioned as necessary, as not all the oral and esophageal secretions will be removed via the nasoesophageal catheter. If the baby requires intubation and ventilation, gentle low pressure ventilation is essential. If the fistula has to be ligated emergently (but is not divided), a delayed primary anastomosis should be performed within 10 days, as recanalization of the distal fistula is likely to occur.
It should be noted that the most important aspect for a thoracoscopic repair is adequate visualization. Impaired visualization often occurs due to the lung being inflated with traditional ventilation. Also, the anesthesiologist (or resident in a teaching hospital) may have limited experience with these cases and fail to understand the importance of low pressure, low volume ventilation to reduce the lung inflation. Moreover, hypercarbia can easily develop. To counteract the problem of lung inflation, CO2 insufflation has been utilized as a positive pressure maneuver to decompress the lung to allow an optimal view. However, this is not always successful in keeping the lung deflated. Over the past 3 years, our group has utilized the high-frequency ventilator (HFV) in the operating room for ventilation in the babies undergoing the thoracoscopic approach. With the use of the HFV and proper positioning of the ETT, the lung remains deflated and an adequate operating space is created. Also, the duration of the operation is not as important because the oscillating ventilator is able to remove the CO2 very efficiently and hypercarbia is not common. Assuming a healthy 3 to 3.5 kg infant, the initial settings are a mean airway pressure of 13 to 15 cm H2O, a δP of 30 to 35, and FiO2 of 70%. This can usually be weaned to 40% or so after 10 to 15 minutes. The major problem with using the HFV is with the movement (shaking) of the baby during the ventilation, but this is something that the surgeon becomes accustomed after the first few cases. The Hertz is usually set initially at 11 to 13 but can be decreased to 8 to 9 if the baby is shaking too much (1 Hz is defined as 60 breaths/s). With this background, we rarely perform these operations on the weekend, as our respiratory therapists are routinely involved with management of the HFV in the operating room.
The baby is positioned supine on the operating table and the oropharynx is suctioned. It is my preference to perform a preoperative bronchoscopy for the purpose of identifying the site of the distal fistula. Others may perform a bronchoscopy to exclude a proximal fistula. By knowing the location of the distal TEF, the surgeon has a better understanding of the gap length between the two esophageal segments and where the distal fistula inserts into the trachea. For instance, if the fistula is identified entering the carina (Fig. 3), the gap length between the distal and proximal segments could be relatively large (up to 3 to 4 cm). On the other hand, if the fistula is seen to enter the mid-trachea, the gap length will likely be much shorter. The distance between the two esophageal segments will also depend on how caudal the proximal esophageal pouch extends. This bronchoscopy takes less than 5 minutes and provides important information to the surgeon performing the repair.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


