Third law of thermodynamics and temperature dependence of heat capacity, enthalpy and entropy
9.1 When and why does a system change?
Having developed the concepts of entropy and enthalpy, we now seek to define a parameter that will tell us which way a reactive system will go: to the right toward products, or to the left to the reactants. We would also like to know what the equilibrium composition of the system will be. We will find that this parameter is the Gibbs energy for a system held at constant temperature and pressure, or the Helmholtz energy for a system held at constant volume and temperature. These energies were defined in Eqs (8.64) and (8.65) and will be derived in the next section.
We will also develop the related concept of chemical potential. The chemical potential plays the role of any other potential. You already know intuitively that in a gravitational potential a body will fall to the lowest possible gravitational potential energy as long as there is no barrier along the pathway. You can put a vase on a shelf. The shelf provides the barrier that keeps the vase from falling to the minimum of gravitational energy (the floor), at least as long as your cat does not intervene. The chemical potential acts in a similar way for a chemical system. Here, we will forget about any potential barriers. Reaction thermodynamics being path-independent tells us nothing about them anyway. We will discuss the formation and presence of such barriers in the chapters on kinetics and dynamics. Here, we concentrate on the way a chemical system develops either in the absence of such barriers or at sufficiently high temperature that any barriers can be overcome without creating bottlenecks.
The change in Gibbs energy is the factor that must be calculated to determine the direction of change of a reaction. Another way of phrasing this is that it is a measure of chemical affinity, and we shall prove that in the next chapter. But Gibbs energy changes are difficult to measure if the only two methods at your disposal are equilibrium concentration measurements and the measurement of voltages coupled with application of the Nernst equation (which we discuss in Chapter 15). These were the only two methods known at the end of the 19th century. Henri le Châtelier (as quoted in Coffey, Cathedrals of Science) stated the importance of determining the Gibbs energy function as follows.
“The determination of the nature of this function would lead to a complete knowledge of the laws of equilibrium. It would permit us to determine a priori, independently of any new experimental data, the full conditions of equilibrium corresponding to a chemical reaction.”
Subsequently, G.N. Lewis developed a method for its calculation but his method could not be applied in practice because a constant of integration could neither be measured nor calculated.
Nernst asserted the Third Law of Thermodynamics, which is what we need to determine the constant that eluded Lewis, based on chemical intuition and extrapolations of Gibbs energy and enthalpy changes. But it was left to Einstein to prove that the Nernst heat theorem as it was called was truly the Third Law of Thermodynamics. Nernst’s heat theorem requires that the heat capacities of solids in their lowest disorder state, which is a perfect crystal, must go to zero at low temperature. Planck, as we will show in Chapter 19, was able to solve the problem of describing the spectrum of thermal black body radiation by introducing his quantum postulate, which states that vibrational energy is quantized. We have used this idea in the development of statistical mechanics. Einstein applied the quantum postulate to the vibrations of a crystal, and the result delivered a very good approximation to the low-temperature behavior of the heat capacity. Debye would later correct Einstein’s model to make it quantitatively accurate over a wider temperature range. Nonetheless, Einstein’s model of heat capacities delivered the proof that was required to turn Nernst’s heat theorem into the Third Law. It also makes clear that the description of the temperature dependence of heat capacities delivers perhaps the most important aspects of understanding chemical thermodynamics. As we shall see, it is from an accurate description of the temperature dependence of heat capacities that we can measure absolute entropies as well as the temperature dependence of both enthalpy and Gibbs energy. We will show shortly that the change in the Gibbs energy upon reaction determines the equilibrium constant. The description of heat capacities provides the bridge that links the quantum mechanical description of energy level structure to thermodynamics.
9.2 Natural variables of internal energy
By combining the First and Second Laws of Thermodynamics, we can express the change in internal energy in terms of T, S, p and V as

This is known as the fundamental equation. This set of variables contains four state variables, as opposed to the path variables δq and δw embodied in the First Law. Therefore, Eq. (9.1) must be applicable to all processes, regardless of whether they are reversible or irreversible.
Equation (9.1) has another interestingly simple property. At constant S, changes in U are related to changes in V by the pressure. That is, a plot of U versus V would have a slope of –p, or in mathematical terms,

Similarly, the internal energy at constant volume but changing entropy changes simply as the temperature,

In this case the partial derivatives have particularly simple forms: they correspond to a state variable (or the negative of a state variable). From this simplicity of form for relating changes of internal energy by way of Eq. (9.1), we call S and V the natural independent variables for the function U. The natural variables are those variables that allow us to determine all other thermodynamic properties by differentiation.
We could, of course, express the internal energy in terms of changes in T and V. Using the appropriate partial derivatives (and results from Eq. (9.1) and another from the Maxwell relations) we find



Thus, upon ‘simplification’ we arrive at a complex function rather than one simply in terms of state variables. Equations (9.1) and (9.6) illustrate that new thermodynamic functions can be defined so as to provide the simplest functional form. Next, we define two new thermodynamic functions of particular importance for determining the natural direction of evolution for a system.
9.3 Helmholtz and Gibbs energies
There are two sets of conditions that are frequently encountered experimentally: constant T and V or constant T and p. Let us now derive new thermodynamic state functions for these two sets of conditions. From the Clausius inequality we have

for any reversible (corresponding to the equal sign) or irreversible process (corresponding to the greater than sign). Remember  . Therefore, at constant volume and no non-pV work, dq = dU, whereas for constant pressure and no non-pV work, dq = dH. Hence, we can develop two parallel derivations, one at constant volume and the other at constant pressure.
. Therefore, at constant volume and no non-pV work, dq = dU, whereas for constant pressure and no non-pV work, dq = dH. Hence, we can develop two parallel derivations, one at constant volume and the other at constant pressure.

We define two new thermodynamic functions

We call the functions the Helmholtz energy A and the Gibbs energy G. At constant temperature, the two state functions change as

Introducing these into the inequalities that follow from the Clausius inequality, we obtain

The choices for these two new state functions were not arbitrary. Instead we see that Eqs (9.14) and (9.15) can be summarized in words as:
For any spontaneous isothermal change, the Helmholtz energy (at constant V) or Gibbs energy (at constant p) must decrease.
A system will always move to a state of lower A or G if a path is available, as shown in Fig. 9.1. This is because a system is always trying to increase the entropy of the universe, that is, the system + the surroundings. Therefore, the condition that defines equilibrium is


Figure 9.1 A spontaneous process is downhill in either the Gibbs energy, dG < 0 at constant pressure and temperature, or Helmholtz energy, dA < 0 at constant volume and temperature. Chemical equilibrium is attained when either the Gibbs or Helmholtz energy has reached a minimum, dG = 0 or dA = 0. The extent of reaction ξ is defined in Chapter 10. In the current example, it is 0 when no reaction has taken place. When the reaction goes to completion, it is 1 mole. When the system settles in to an equilibrium, ξ has some intermediate value.
9.3.1 Example
How much work can be extracted from glucose metabolism performed reversibly at 25 °C given that ΔrU°m = −2801.3 kJ mol−1 and ΔrS°m = 260.7 J K−1 mol−1?
Treat the body as a constant volume system. The Helmholtz energy is given by
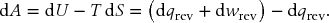
Here we have used the first law to substitute for dU. In the second term, we used dqrev = T dS. Hence, dA = dwrev or

for a finite process. Therefore, the change in the Helmholtz energy is the maximum amount of work that can be extracted from a system at constant temperature and volume. Now calculate the change in Helmholtz energy

9.4 Standard molar Gibbs energies
A balanced stoichiometric chemical reaction is written with stoichiometric coefficients a, b, and so forth, which are all positive numbers

The IUPAC defines stoichiometric numbers νA, νB, etc. as signed quantities, which are negative for reactants and positive for products. Thus, for the equation written above, νA = −1, νB = −2, νY = 3, νZ = 4.
Equation (9.11) applies to each substance in a chemical system; therefore changes in the Gibbs energy can be calculated from the changes in enthalpy and entropy that accompany the reaction. Standard entropies and enthalpies of reaction are combined to obtain the standard Gibbs energy of reaction according to

In a standard formation reaction there is one and only one product and it has a stoichiometric coefficient of unity. The reactants are elements in their standard state. We can relate the change in Gibbs energy of any reaction to the Gibbs energies of formation of reactants and products. We multiply the Gibbs energy of formation of each species by the corresponding stoichiometric coefficient. We then sum all the values of the products, sum all the values of the reactants, and then subtract the reactant sum from the product sum,

More formally correct, we use the fact that the stoichiometric number of the jth species is a signed quantity to write this as

By convention, ΔfG° for an element in its standard state is zero at all temperatures. Similar relationships are used to relate reaction enthalpy and entropy changes to enthalpies of formation and standard entropies


Whereas ΔfH° for an element in its standard state is also defined as zero at all temperatures, we shall see below that according to the Third Law of Thermodynamics absolute entropies S° are zero for a perfect crystal at absolute zero.
9.4.1 Example
Determine ΔrG°m (298 K) for the combustion of C2H5OH given that ΔfG°m (298 K) for C2H5OH(l), H2O(l) and CO2(g) are –175, –237 and –394 kJ mol−1, respectively.
First, write a balanced chemical reaction

Then apply this stoichiometry to Eq. (9.20)

The ‘per mole’ in the Gibbs energy change refers to ‘per mole of the reaction as written,’ which in this case happens to be per mole of ethanol combusted but per 3 mol O2 reacted.
9.4.2 Directed practice
How many products and how much product is made in a standard formation reaction?
9.5 Properties of the Gibbs energy
How does the Gibbs energy change with temperature and pressure? As for any function, we can write this dependence abstractly in terms of partial differential equations

We now evaluate the coefficients represented by the partial derivatives and their functional form. The Gibbs energy is defined by

The molar Gibbs energy is a state property (path independent) as are Hm, T, and Sm. For infinitesimal changes, we obtain by differentiation

By differentiating the definition of enthalpy, Hm = Um + pVm, we know that

For a closed system doing no non-expansion work, we can substitute for dUm from the fundamental equation, Eq. (9.1) above. We then substitute for dHm in Eq. (9.25) from Eq. (9.26) to find

Therefore, Gm is a function of p and T and the form of this function is given by the partial derivatives

and

Because Sm > 0 for all substances, Gm always decreases with increasing T (at constant pressure and composition), as shown in Fig. 9.2. Larger changes in Gm occur with larger values of Sm; therefore, Gm is more sensitive to T changes when gases are involved in the process than when condensed phases are the only ones present.

Figure 9.2 The molar Gibbs energy is a decreasing function of temperature at constant pressure. As the molar entropy is of greater magnitude for gases than that of liquids and solids (away from the critical point), the effect of temperature is much greater for gases than condensed phases.
Similarly Vm > 0 for all substances. Therefore, Gm increases when p increases, as shown in Fig. 9.3. The molar Gibbs energy is more sensitive to changes in p for gases than for condensed phases.
9.5.1 Gibbs–Helmholtz equation
From the dependence of G on T in Eq. (9.28) and the definition of G in Eq. (9.11)

We shall see later that the equilibrium constant K is related to ΔG°m/T by

Therefore by determining how G/T changes with temperature, we can also determine how K changes with T. Using the chain rule to differentiate G/T, we obtain

A useful result to remember is

Substituting into Eq. (9.32) from Eq. (9.30) we obtain the differential form of the Gibbs–Helmholtz equation

Sometimes written as

That is, a plot of G/T versus 1/T has slope H. The same result holds for ΔG with ΔH in place of H. Thus, by integrating Eq. (9.35) we can find the value of ΔG at any temperature T2 from a value at a reference temperature T1 according to
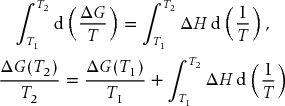
If ΔH is constant it can be pulled from the integral to yield the integrated form of the Gibbs–Helmholtz equation

9.5.1.1 Directed practice
Derive the Gibbs–Helmholtz equation in Eq. (9.34) by substituting Eq. (9.30) into Eq. (9.32).
9.5.1.2 Example
Calculate the change in molar Gibbs energy if the pressure is increased from 1.0 bar to 2.0 bar at 298 K for (a) liquid water (incompressible liquid) (b) water vapor (ideal gas).
The molar Gibbs energy depends on changes in pressure and temperature according to

The processes are isothermal, therefore dT = 0 and
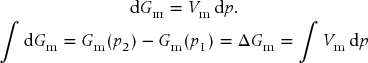

For the incompressible liquid, the molar volume is a constant and can be pulled from the integral. For the ideal gas, we must substitute explicitly for the molar volume and integrate. The change is positive in both cases but about 1000-fold larger for the gas.
9.5.2 Gibbs energy of an ideal gas at arbitrary pressure
If the initial pressure is taken to be the standard state pressure, p° = 1 bar, we may write

In other words, taking the molar Gibbs energy of an ideal gas at the standard state pressure as the standard molar Gibbs energy G°m, then the Gibbs energy of an ideal gas at any pressure p is related to the standard state value plus the RTln (p/p°) term.
9.5.3 Gibbs energy and electrical work
Equation (9.1) is valid if only pV work is done. In an electrochemical system electrons are generated and electrical work wel is done. Consequently, we have to modify the fundamental equation to include this term

Therefore

At constant p and T for a finite process,

Hence, ΔG is the maximum nonexpansion (e.g., electrical) work that can be done by the system at constant p and T.
9.5.3.1 Example
In a methane fuel cell, CH4 undergoes the same redox reaction as in the combustion process to produce CO2 and H2O and generates electricity. Calculate the maximum electrical work that can be obtained from 1 mol of CH4 at 25 °C.
Start with a balanced reaction
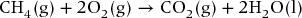
Using tables to find enthalpies of formation ΔfH°m and standard molar entropies S°m, ΔrH°m = −890.3 kJ mol−1 and ΔrS°m = −242.8 J K−1 mol−1 are calculated.

The work done is less than the heat generated. This is because of the unfavorable entropy change. If the enthalpy of combustion were used to do work in a heat engine, then the efficiency would be limited, as in a Carnot engine. However, in theory, 100% of the Gibbs energy released in a fuel cell is available for work. Therefore, a fuel cell is in principle more efficient than any internal combustion engine because it is not limited by the Second Law.
9.6 The temperature dependence of ΔrCp and H
Let us assume that in changing temperature from T1 to T2, none of the reactants or products in a reaction undergoes a phase change. Enthalpy H is a state function defined by

Enthalpy is a state function that depends on p and T according to

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


