, Arthur H. Cohen2, Robert B. Colvin3, J. Charles Jennette4 and Charles E. Alpers5
(1)
Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee, USA
(2)
Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, California, USA
(3)
Department of Pathology Harvard Medical School, Massachusetts General Hospital, Boston, Massachusetts, USA
(4)
Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, North Carolina, USA
(5)
Department of Pathology, University of Washington, Seattle, Washington, USA
Abstract
Classical Alport syndrome is an X-linked disease and is the most common form of Alport syndrome (90 % of patients), with an overall incidence of Alport syndrome in the United States of 1:5,000 to 1:10,000 [1–4]. Hematuria develops in early childhood in all male patients with X-linked Alport and in 95 % of female carriers of X-linked Alport and in nearly all autosomal recessive carriers. Affected males with X-linked Alport develop progressive hearing loss in one third and ocular defects and progression to renal failure in 30–40 % by early adulthood with more than 90 % with end-stage renal disease by age 40. Eye defects develop in 20–30 % of males with X-linked Alport, among which anterior lenticonus is the most common lesion. A large study of heterozygous female carriers of X-linked Alport syndrome demonstrated development of proteinuria in about 75 and 30–40 % with end-stage renal disease after age 60.
Alport Syndrome
Introduction/Clinical Setting
Classical Alport syndrome is an X-linked disease and is the most common form of Alport syndrome (90 % of patients), with an overall incidence of Alport syndrome in the United States of 1:5,000 to 1:10,000 [1–4]. Hematuria develops in early childhood in all male patients with X-linked Alport and in 95 % of female carriers of X-linked Alport and in nearly all autosomal recessive carriers. Affected males with X-linked Alport develop progressive hearing loss in one third and ocular defects and progression to renal failure in 30–40 % by early adulthood with more than 90 % with end-stage renal disease by age 40. Eye defects develop in 20–30 % of males with X-linked Alport, among which anterior lenticonus is the most common lesion. A large study of heterozygous female carriers of X-linked Alport syndrome demonstrated development of proteinuria in about 75 % and 30–40 % with end-stage renal disease after age 60.
Alport syndrome is due to mutations of collagen type IV [3–6]. The organs affected reflect the sites where collagen IV is crucial for function. Collagen type IV is made up of heterotrimers of alpha chains. These six alpha chains are encoded by genes arranged in pairs on three different chromosomes: COL4A1 and COL4A2 are on chromosome 13, COL4A3 and COL4A4 are on chromosome 2, and COL4A5 and COL4A6 are on the X chromosome. The mutation in the classic form of Alport occurs in the α5 (IV) collagen chain (COL4A5). The autosomal recessive form accounts for most of the remaining patients and is due to mutations in both alleles of α3 or α4 type IV collagen genes (COL4A3 or COL4A4). Rare cases of autosomal dominant Alport due to heterozygous mutations in COL4A3 or COL4A4 also occur, with a highly variable clinical course and reduced penetrance [7]. Alport syndrome and coexisting diffuse leiomyomatosis are linked to large gene deletions that span the adjacent 5′ ends of the adjacent COL4A5 and COL4A6 genes [5].
Pathologic Findings
Light Microscopy
There are no significant light microscopic abnormalities early in the disease [1]. At later stages, glomerulosclerosis, interstitial fibrosis, and prominent interstitial foam cells, nonspecific and just indicative of proteinuria, are typical. Glomeruli show varying stages of matrix expansion and sclerosis.
Immunofluorescence Microscopy
Standard immunofluorescence (IF) may show nonspecific trapping of immunoglobulin M (IgM) in the mesangium. Special IF studies for subtypes of type IV collagen on either skin or renal biopsy may be helpful in distinguishing between causes of thin glomerular basement membrane (GBM), which may be the only lesion in early Alport, the carrier state for X-linked Alport, or the so-called benign familial hematuria (see below) [3, 5, 8–11].
When any one of the three alpha chains, α3, α4, or α5, is mutated, the normal collagen IV (α3, 4, 5) heterotrimer cannot form. In kidney biopsies, about 70–80 % of males with X-linked Alport, where α5 is mutated, thus lack staining of the GBM, distal tubular basement membrane, and Bowman’s capsule for α3, α4, and α5 (IV) chains [3, 10, 11]. In autosomal recessive Alport, where α3 or α4 is mutated, the GBMs usually show no immunostaining for α3, α4, or α5, because there is an inability to form the normal α3, α4, or α5 type IV collagen heterotrimer of the GBM. In these autosomal recessive cases, in contrast to X-linked cases, α5 remains normally expressed in Bowman’s capsule and distal tubular basement membrane, because the α(1,1,2)/(5,5,6) heterotrimers can still be assembled in these patients. Female heterozygotes for X-linked Alport syndrome frequently show mosaic staining of GBM and distal TBM for α3, α4, and α5 (IV) chains and skin mosaic staining for α5 (IV). Patients with autosomal dominant Alport have not been studied immunohistochemically. Skin biopsies are thus only useful in diagnosis of classic X-linked Alport. In these affected patients, skin biopsy often shows absent staining for α5 type IV collagen. In contrast, α3 and α4 are not normally expressed in the skin, so this approach is not useful for diagnosis of recessive Alport [10, 12].
Of note, some cases with Alport syndrome clinically and by renal biopsy showed apparent normal α5 type IV immunostaining pattern. About 20 % of male classic Alport patients and affected homozygous autosomal recessive Alport patients show faint or even normal immunostaining of the skin or GBM for α5 [3]. This is postulated to reflect a mutation that results in protein that albeit abnormal still expresses the epitope recognized by the available antibodies. Thus, the absence or mosaic staining of α5 type IV in the biopsy is helpful in indicating a basement membrane abnormality, but an apparent normal staining pattern in either skin or kidney does not definitively rule out Alport syndrome [10, 11]. Further, many patients with apparent benign familial hematuria have thin basement membranes and are carriers of autosomal recessive Alport with normal immunostaining (see below).
Electron Microscopy
The diagnostic lesion consists of irregular thinned and thickened areas of the GBM with splitting and irregular multilaminated appearance of the lamina densa, the so-called basket weaving (Fig. 7.1) [2]. In between these laminas, granular, mottled material is present. In boys with early-stage classic Alport syndrome, the GBM may show only thinning rather than thickening. Female carriers of the COL4A5 mutation also show only thin basement membranes, as do carriers of the autosomal recessive form of Alport. The GBM thickness normally increases with age [13–15]. Normal thickness in adults in one series was 373 ± 42 nm in men versus 326 ± 45 nm in women. Glomerular basement thickness <250 nm in adults has been used as a cutoff for diagnosis in many series. Of note, thinning of GBMs should be a diffuse lesion, affecting more than half the extent of the GBM, to consider a diagnosis of thin GBM and its differential. In children, the diagnosis of thin basement membranes must be made with caution, establishing normal age-matched controls within each laboratory. In our laboratory, we found a range of GBM thickness in normal children from approximately 110 nm at age 1 year to 222 ± 14 nm in 7-year-olds.
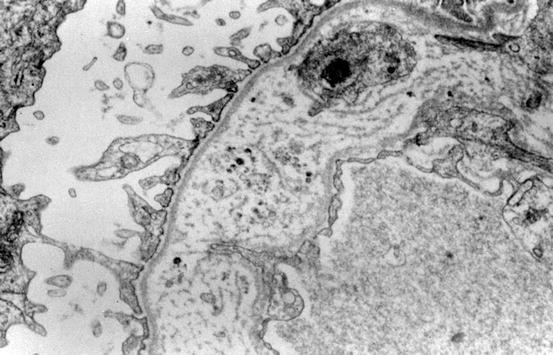
Fig. 7.1
The glomerular basement membrane (GBM) is thickened with “basket-weaving” appearance, diagnostic of Alport syndrome (electron microscopy)
Etiology/Pathogenesis
Alport syndrome results from the inability to form normal type IV collagen heterotrimers. When α5 (or α3 or α4) is mutated, there is an inability to form the normal heterotrimers of the GBM. The organs involved in Alport syndrome reflect sites where these type IV collagen chains are normally expressed and are essential for function, namely, the kidney, eye, and ear. In the kidney, heterotrimers of α3, α4, and α5 type IV collagen are expressed in the GBM, whereas α(1, 1, 2)/(5, 5, 6) heterotrimers are expressed in Bowman’s capsule and in some tubular basement membranes [5]. At birth, α(1,1,2) heterotrimers are normally present in the immature glomerulus in the GBM, with gradual shift to the mature expression pattern. In the normal adult, α(1,1,2) remains expressed in the mesangium and also in Bowman’s capsule. The switch to normal adult α(3,4,5) heterotrimers in the GBM cannot occur in Alport due to mutation in one of these chains.
The mechanism(s) of progressive renal scarring in Alport syndrome is unknown. In a report of seven patients with Alport syndrome, decreased proteinuria occurred in response to angiotensin-converting enzyme inhibitor (ACEI), and after an initial decrease of the glomerular filtration rate (GFR), renal function increased toward the starting levels by 24 months [16]. Larger retrospective analyses have shown benefit of angiotensin inhibition starting at early stages of disease [17]. Female carriers of X-linked Alport syndrome have variable outcomes. About 75 % of female carriers developed proteinuria, a risk factor for their eventual progression to end-stage kidney disease. About 30–40 % developed end-stage kidney disease by age 60, and about a quarter developed deafness [18].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


