, Arthur H. Cohen2, Robert B. Colvin3, J. Charles Jennette4 and Charles E. Alpers5
(1)
Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee, USA
(2)
Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, California, USA
(3)
Department of Pathology Harvard Medical School, Massachusetts General Hospital, Boston, Massachusetts, USA
(4)
Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, North Carolina, USA
(5)
Department of Pathology, University of Washington, Seattle, Washington, USA
Abstract
Hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) share the morphologic lesion of thrombotic microangiopathy (TMA), characterized by thrombi occluding the microvasculature. The HUS and TTP syndromes overlap clinically [1–9]; however, there are differing pathogeneses (see below). TTP is more common in adults and is characterized by fever, bleeding, hemolytic anemia, kidney injury, and neurologic impairment. Hemolytic uremic syndrome is characterized by acute kidney injury, nonimmune hemolytic anemia, and thrombocytopenia, and it is most common in infants and small children. The renal manifestations at presentation include hematuria and low-grade proteinuria with elevated creatinine in severe cases. Intravascular hemolysis is evident by increased bilirubin and lactate dehydrogenase (LDH), reticulocytosis, and low haptoglobin. Both HUS and TTP cause thrombocytopenia. In our experience, and that of others, the peripheral blood manifestations may not be detected by the time a renal biopsy is performed, especially in the transplant setting [10].
Introduction/Clinical Setting
Hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) share the morphologic lesion of thrombotic microangiopathy (TMA), characterized by thrombi occluding the microvasculature. The HUS and TTP syndromes overlap clinically [1–9]; however, there are differing pathogeneses (see below). TTP is more common in adults and is characterized by fever, bleeding, hemolytic anemia, kidney injury, and neurologic impairment. Hemolytic uremic syndrome is characterized by acute kidney injury, nonimmune hemolytic anemia, and thrombocytopenia, and it is most common in infants and small children. The renal manifestations at presentation include hematuria and low-grade proteinuria with elevated creatinine in severe cases. Intravascular hemolysis is evident by increased bilirubin and lactate dehydrogenase (LDH), reticulocytosis, and low haptoglobin. Both HUS and TTP cause thrombocytopenia. In our experience, and that of others, the peripheral blood manifestations may not be detected by the time a renal biopsy is performed, especially in the transplant setting [10].
Pathologic Findings
Light Microscopy
Fibrin and platelet thrombi are present, primarily in the glomeruli [1–4]. Fibrin is best visualized on hematoxylin and eosin or silver stains. Lesions may extend to arterioles, with some overlap with progressive malignant hypertension and scleroderma, where arteriolar and even larger vessel involvement occurs (Figs. 11.1 and 11.2). Mesangiolysis occurs frequently but is a focal, subtle lesion that may be overlooked [11]. Mesangial areas seem to “unravel,” resulting in very long, sausage-shaped capillary loops due to the loss of mesangial integrity and coalescence of adjoining loops.
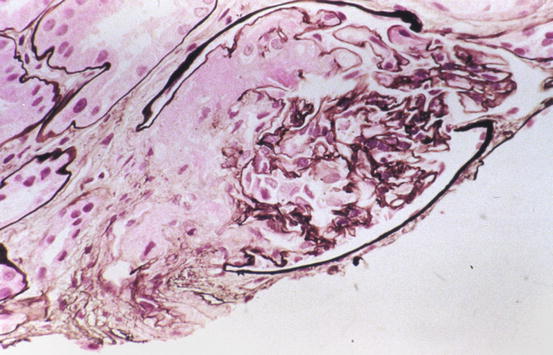
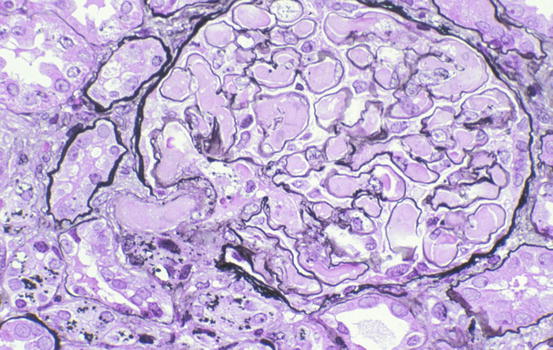
Fig. 11.1
Segmental red blood cells (RBCs) and fibrin in capillary loops and arteriole in glomerulus in thrombotic microangiopathy (Jones silver stain)
Fig. 11.2
Entire glomerulus and arteriole are filled with chunky, eosinophilic fibrin in this case of hemolytic uremic syndrome (HUS) (Jones silver stain)
In infants and young children, thrombotic lesions predominate [4]. In older children and adults, varied lesions occur. Many glomeruli may show only ischemic changes with corrugation of the glomerular basement membrane and retraction and collapse of the glomerular tuft. Segmental glomerular necrosis may be seen with rare well-developed fibrin thrombi. Arterioles and arteries, when involved, show thrombosis and sometimes necrosis of the vessel wall, with intimal swelling, mucoid change, and intimal proliferation. Fragmentation of red blood cells within the vessel wall may also be present. Tubular and interstitial changes are proportional to the degree of glomerular changes. In severe cases, cortical necrosis can occur [12].
Secondary changes late in the course include glomerular sclerosis, either segmental or global. Reduplication of the glomerular basement membrane may occur in the late phase due to organization following endothelial injury.
Immunofluorescence Microscopy
Immunofluorescence studies show no immunoglobulin deposits. Complement and immunoglobulin M (IgM) may be present in sclerotic areas. Fibrin and fibrinogen are present in affected glomeruli and arterioles.
Electron Microscopy
Endothelial cells are frequently swollen, and detachment may be seen by electron microscopy. Fibrin tactoids may be present in affected glomeruli (Fig. 11.3). Mesangiolysis is a prominent finding in early phases [11].
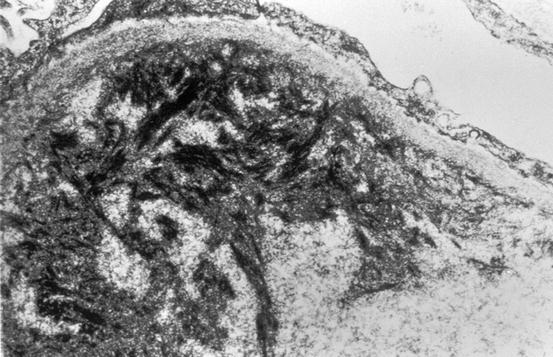
Fig. 11.3
Fibrin tactoids in subendothelial area in thrombotic microangiopathy (electron microscopy)
In the subacute and chronic phase, the increased lucency of the lamina rara interna is in part correlated to breakdown of coagulation products (Fig. 11.4). This zone contains breakdown products of fibrin, laminin, and fibronectin [2].
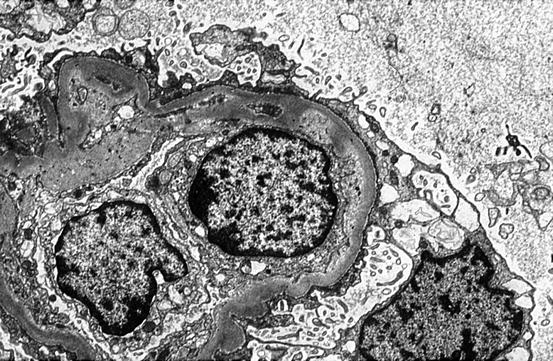
Fig. 11.4
Increased lucency of lamina rara interna and glomerular basement membrane (GBM) corrugation in HUS (electron microscopy)
Etiology/Pathogenesis
Thrombotic microangiopathy is the key lesion present in both HUS and TTP. Numerous etiologies are recognized (Table 11.1) [7, 13, 14]. HUS/TTP has also been classified as diarrhea associated or not, D+ or D−. The typical diarrhea-associated (D+) form of HUS accounts for the vast majority of HUS cases and is most often associated with Shiga-like toxin or verotoxin [4, 9, 12]. Most of these infections are due to the Escherichia coli serotype O157:H7. Verotoxin was associated with ~90 % of cases of HUS in children in North America and Europe. Undercooked hamburger meat is most closely associated with such outbreaks in North America, pointing to cattle as an important reservoir for the implicated E. coli serotype O157:H7. In addition, this E. coli strain can be transmitted from person to person, and outbreaks associated with swallowing contaminated lake water or ingestion of contaminated fruit or vegetables or cider have occurred. In a recent outbreak, Shiga-toxin-producing E. coli O104:H4 was identified and linked to contaminated sprouts, and most patients were adult [15].
The mature verotoxin has alpha and beta subunits. The beta subunits interact with the target cell, most often the endothelial cell, binding to the glycolipid Gb3 protein. The alpha unit is cleaved and taken up by endocytosis, inactivating 60S ribosomes, thereby causing cell death. The Gb3 receptor for verotoxin is highly expressed in human kidney, perhaps underlying the susceptibility of the kidney to this toxin [16]. However, Gb3 levels were not different in normal children vs. adults, so the excess risk of children for D+ HUS cannot be simply explained by overexpression of Gb3 [17].
D(−) HUS, also called atypical HUS (aHUS), comprises about 10 % of cases of HUS/TTP. With atypical HUS (D–) no diarrheal prodrome is seen, and Shiga-like toxin is not identified. About half of these patients have an underlying genetic abnormality of key regulatory molecules of the complement cascade, such as factor H (CFH), factor I (CFI), or membrane cofactor protein (MCP) [18, 19]. Ongoing activation of complement injures the endothelium and thrombosis ensues. Disease has early onset, before 1 year of age with CFH and CFI defects, and is often relapsing and leads to end-stage kidney disease. In some patients there may be an autoantibody to factor H, with underlying defect of factor H, the so-called DEAP-HUS (deficient for CFHR proteins and factor H autoantibody positive) [19]. Plasmapheresis may be beneficial in these patients. Liver and kidney transplant may be needed for patients with defects in CFH or CFI, which are synthesized in the liver, whereas patients with defect of MCP, a factor synthesized systemically, may generate enough factor from kidney transplant. Eculizumab, a humanized monoclonal antibody against complement protein C5 that thus inhibits activation of the terminal complement pathway, has been used to treat the complement dysregulation [15].
Familial TTP is most often due to constitutional deficiency of a von Willebrand factor (vWF)-cleaving protease, whereas a nonfamilial form of TTP seems to be caused by an acquired inhibitor of this protease. This protease is now called ADAMTS13 (a member of the “a disintegrin and metalloprotease with thrombospondin type 1 repeats” family of zinc metalloproteases) [7]. The long vWF multimers activate platelets and cause thrombosis when ADAMTS13 function is defective. However, there is overlap with varying phenotypes of injury even within the same family and overlap of the HUS-TTP spectrum.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


