INTRODUCTION
Blood moves through the vascular system secondary to pressure and osmotic gradients similar to a plumbing system, but, unlike our household pipes, the system of blood vessels is not made up of static and inflexible structures. The vascular system is dynamic and always changing to regulate blood flow to the body and its essential organs. The system of blood vessels with the heart at its center is critical for delivering oxygenated blood and essential nutrients to organs and tissues and removing and transporting waste products.
This chapter will cover the normal circulatory system and its structures and congenital and acquired diseases including atherosclerosis, trauma, and inflammatory and infectious processes. The pulmonary vascular system and its diseases will also be discussed.
EMBRYOLOGY OF BLOOD VESSELS
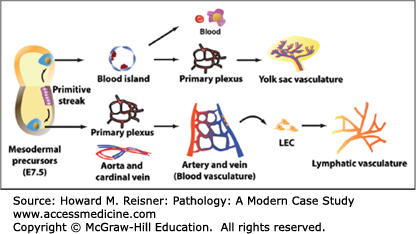
The cardiovascular system develops very early in gestation (as early as 15–16 days) due to the limited energy sources available from the egg and yolk sac. The cardiovascular system delivers nutrients to the increasingly actively dividing cells and disposes of waste products through its connections with the maternal vasculature of the placenta. The heart and blood vessels are created from the mesoderm that forms blood islands (isolated cell masses) around which the endothelial tubes are formed. Vascular smooth muscle cells and connective tissue derived from the local mesoderm then surround the endothelial tubes. The heart begins to beat and blood begins to circulate throughout the blood vessel network around the 4th week of gestation (Figure 7-1).
FIGURE 7-1
Embryological development of blood vessels. In the extraembryonic yolk sac, mesodermal precursor cells aggregate to form blood islands, the sites of development of endothelial and primitive blood cells. Within the blood islands, centrally-located cells become primitive blood cells, whereas outer cells give rise to endothelial cells (ECs). ECs then form the vascular primary plexus which is subsequently remodeled to form the yolk sac vasculature. In the embryo proper, mesodermal precursor cells differentiate into the vascular primary plexus and major vessels, aorta, and cardinal vein. After arterial and venous ECs are specified, the complex blood vasculature is formed via extensive remodeling. At embryonic day (E) 9.5, a subset of ECs of the cardinal vein acquires a lymphatic endothelial cell (LEC) fate and develops into lymphatic vessels. From Park C, Kim TM, Malik AB.Transcriptional regulation of endothelial cell and vascular development. Circ Res. 2013;112:1380–1400.
QUICK REVIEW Normal Anatomy of Blood Vessels
Blood vessels consist of arteries, veins, and capillaries that are categorized according to their structure, size, and function. The arterial system carries oxygenated blood away from the heart to the organs and periphery providing oxygen and nutrients to the cells at a distance. The capillary system is responsible for the exchange of new supplies and the waste products. The venous system carries the deoxygenated blood with its waste products back to the heart for transport to the lungs for replenishment of oxygen. Lymphatics are a back-up system that operates to assist the vascular system.
Arteries are made up of three layers: the intima, media, and adventitia. The intima is composed of a confluent, single-cell layer of endothelium lining the blood vessel lumen, a basement membrane, and subendothelial connective tissue along the luminal side of the internal elastic lamina. The cells of the intima derive their nutrients and oxygen directly from the luminal contents. The media is the middle and thickest layer of the artery bounded by the internal and external elastic laminae. Its components include smooth muscle fibers and collagen. Additional components (elastic fibers) vary depending on the size and function of the artery. Changes in the arterial media are due to local adaptations for mechanical or metabolic needs of the organ or tissue. These adaptive structural changes are primarily seen in the media and external elastic lamina. The outermost layer, the adventitia, is composed of the external elastic lamina, connective tissue, nerves, and small blood vessels (vasa vasorum). The vasa vasorum are the “artery’s artery” and are the source of nutrients and oxygen for the arterial media.
The arterial system, responsible for conducting the blood to the periphery and for creating peripheral resistance, is divided into three types based on size, structural features, and function. The three arterial types are divided by their medial components and include elastic arteries, muscular arteries, and small arteries and arterioles.
Elastic arteries are the large-diameter, low-resistance arteries of the body and include the aorta (the largest blood vessel in the body), aortic branches (the brachiocephalic, subclavian, common carotids, and iliac arteries), and the pulmonary arteries. The media of the elastic arteries is rich in elastic fibers arranged in compact layers that alternate with smooth muscle cells. The elastic component allows for expansion of the artery during cardiac systole (contraction of the heart), creating kinetic energy for elastic recoil and propulsion of blood to the periphery during cardiac diastole (relaxation of the heart). The media of the elastic arteries is supplied with oxygenated blood and nutrients through the small arteries throughout the adventitia.
The muscular, medium-sized arteries are considered the distribution system and conduct the blood to the organs and periphery. The major muscular arteries to be discussed include the coronary, renal, and femoral arteries. The media of these arteries is composed of a circular to spiral arrangement of smooth muscle cells without elastic fibers. The lack of elastic fibers allows more efficient contraction of the artery in response to appropriate stimulation. The internal and external elastic laminae are the only elastic elements of the muscular arteries. As the muscular arteries branch, the media thins, the internal elastic lamina disappears, and the intima is a single layer of endothelial cells (ECs).
Small arteries and arterioles are differentiated by size: small arteries are <2 mm and arterioles are 20 to 100 μm in diameter. They are made up of a single-cell layer of endothelium, a few layers of smooth muscle cells, and adventitia. Small arteries and arterioles are critically important as they regulate regional blood flow and systemic blood pressure by changes in the caliber of the lumen.
The small arteries and arterioles lead to the capillary system where exchange of nutrients, oxygen, and waste products actually occurs through the smallest (diameter of a red blood cell), thin-walled (single endothelial layer supported by scattered smooth muscle cells) vessels. The capillary system has a markedly increased surface area and decreased flow velocity to maximize the exchange between the blood and tissues. The highest density capillary systems are found in organs and tissues with a high metabolic rate (myocardium). ECs are semipermeable allowing for exchange controlled by molecular size and charge of the substrate. Like other portions of the vascular system, capillaries are modified according to the tissue or organ they supply by alterations of their ECs creating changes in permeability and their ability to transport substances across cell membranes (pinocytosis). For example, fenestrated capillaries in renal glomeruli are specifically adapted to allow filtration of plasma.
From the capillary system, the blood begins its return to the heart through postcapillary venules (small veins), to collecting venules, to small veins, medium veins, and large veins. Veins are thin-walled, large–volume, and large-diameter vessels with less-organized walls. They are also surrounded by less support allowing for irregular dilatation, compression, and penetration by tumor and inflammatory cells. Collectively, the venous system contains approximately two-thirds of our blood volume at one time. Reverse flow is prevented by venous valves.
Lymphatics are thin-walled, endothelial-lined channels that serve as a back-up for the vascular system by collecting interstitial fluids and returning them to the vascular system via the thoracic duct. Of note, lymphatics are very important as they are easily accessed by tumor cells and microorganisms leading to dissemination of disease.
The pulmonary vasculature is a crucial part of the cardiovascular system as it carries deoxygenated blood from the right side of the heart to the lungs. If this pathway is disrupted or interrupted, the blood can no longer reach the lungs for oxygenation leading to death.
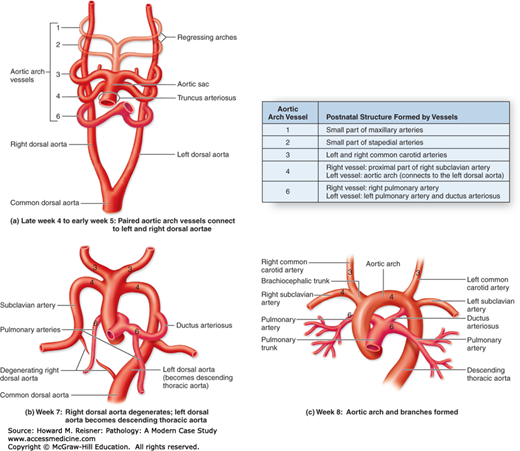
Embryologically, the aorta and main pulmonary artery are formed from the truncus arteriosus (the single blood vessel arising from the heart) by septation. Therefore, prior to birth, the main pulmonary artery is a large elastic artery similar to the aorta. After birth, its elastic fibers become less organized (Figures 7-2 and 7-3).
FIGURE 7-2
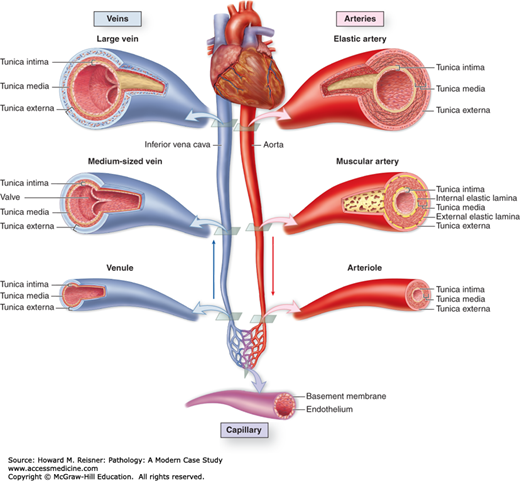
Vessels of the blood circulatory system. The heart is the principal organ of the blood circulatory system, pumping blood throughout the body and providing one of the forces by which nutrients leave the capillaries and enter tissues. Large elastic arteries leave the heart and branch to form muscular arteries. These arteries branch further and enter organs, where they branch further to form arterioles. These arterioles branch into the smallest vessels, the capillaries, composed by a single cell layer allowing exchange of nutrients and waste products between blood and surrounding tissue. Capillaries then merge to form venules, which merge further into small- and later medium-sized veins. These veins leave organs and form larger veins that eventually bring blood back to the heart. From Mescher AL. Junqueira’s Basic Histology Text & Atlas. 12th ed. New York, NY: McGraw-Hill Lange; 2010. Figure 11-1, page 186.
DISEASES OF THE VASCULAR SYSTEM
Congenital vascular anomalies are most commonly vascular structures with abnormal branching and anastomotic connections that are developed in utero. However, such congenital abnormalities are not necessarily genetically transmitted. Two important congenital vascular anomalies include the berry aneurysm and the arteriovenous malformation (AVM).
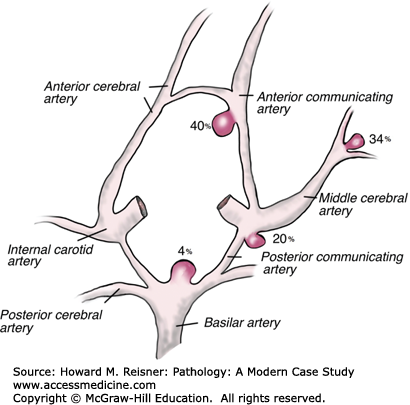
The berry aneurysm is a saccular aneurysm (a dilated outpouching of a blood vessel that forms a sac-like structure with its artery at its base) found in approximately 2% of autopsies and in 3% to 6% of the general population. The abnormality is most commonly found intracranially. At birth, the arterial medial layer is abnormal creating a focal weakness in the arterial wall. The dilated outpouching is not present at birth, but forms over time with increased vascular pressure and various risk factors including cocaine use and cigarette smoking. The majority (90%) of berry aneurysms are found in the anterior cerebral circulation of the circle of Willis at its major branch points (internal carotid arteries, anterior communicating arteries, etc.), where there is significant turbulence and shear forces. Multiple aneurysms in a single person are seen in 20% to30% of cases. Annual rupture rates in one large study showed that aneurysms <10 mm had a rupture rate of 0.05%. Rates of rupture were higher (~1%) for aneurysms >10 mm in diameter, those that were symptomatic, or those located in the posterior circulation.
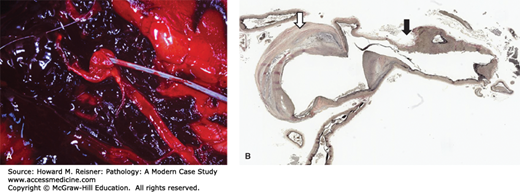
Most aneurysms are clinically silent and asymptomatic, remaining undetected until rupture. Clinical presentation of a ruptured berry aneurysm (the first presentation in over 50% of cases) is most commonly “the worst headache of my life” with nausea and vomiting. There may be a loss of consciousness. The ruptured aneurysm leads to a diffuse subarachnoid hemorrhage (blood over the surface of the brain below the arachnoid) (Figure 7-4A). The hemorrhage leads to contraction of cerebral vasculature (vasospasm) and decreased blood flow, which leads in turn to decreased oxygenation of the brain or cerebral ischemia. Rupture of a cerebral aneurysm is a medical emergency due to the significant morbidity and mortality. With the first rupture, 25% to 50% of patients will die. Some patients may regain consciousness while others will not.
FIGURE 7-4
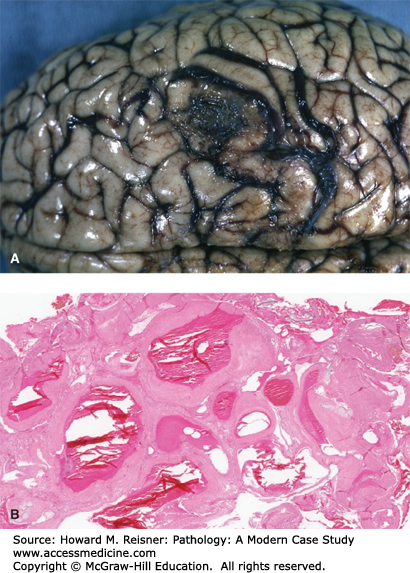
(A) Acute subarachnoid hemorrhage secondary to ruptured berry aneurysm: probe indicates ruptured saccular aneurysm of middle cerebral artery. Note the large thrombus on the left of image. From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill. (B) Berry aneurysm (elastic tissue stain). Area of rupture is shown by a black arrow. Area of dilatation and disorganization of elastic tissue is shown by a hollow arrow. From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
Gross pathology of the aneurysm is a sac-like structure protruding from its artery. Histologically, the aneurysm wall is made up of a thickened, hyalinized intimal layer and adventitia. The media in the area of the aneurysm is absent (Figure 7-4B). The frequency of berry aneurysms varies by anatomic location (Figure 7-5).
While the majority of berry aneurysms are sporadic and not genetically transmitted, they may be associated with polycystic kidney disease (autosomal dominant polycystic kidney [ADPK] disease), Ehlers–Danlos and Marfan syndromes, neurofibromatosis type I, fibromuscular dysplasia, and coarctation of the aorta.
AVMs (also known as arteriovenous fistula) are abnormal direct communication between arterial and venous vessels without the interposed, normal capillary system leading to a short circuit of blood flow. These abnormalities are more commonly found in men (2:1) and are most likely to be clinically recognized between 10 and 30 years of age. They may present as a new-onset seizure disorder with stroke-like symptoms or rupture.
CASE 7-1
A previously healthy, 30-year-old woman complained of the “worst headache” of her life before collapsing at an aerobics class. She was rushed to the local emergency department where it was found on CT scan that she had diffuse subarachnoid hemorrhage. An autopsy showed a ruptured cerebral berry aneurysm (Figure 7-4)
Grossly, the AVM appears as worm-like channels. Histologically, the enlarged blood vessels show abnormal arteries and veins (Figure 7-6).
FIGURE 7-6
Arteriovenous malformation of the brain. (A) Gross fixed tissue, external cortical view. From Peter Anderson D.V.M., Ph.D., PEIR Digital Library Image 9386. (B) Microscopic view (low power). From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
An abnormal, localized, usually superficial collection of capillaries commonly seen in infancy or childhood (with a frequency of 1:200) is termed a capillary hemangioma (Figure 7-7A). A number of these collections are present at birth (30%) and may enlarge with a child’s growth. They are most often seen on the head or neck, but up to one-third can be found in the liver, spleen, or kidneys. These collections do not undergo malignant transformation. Most (75%–90%) will regress by 7 years of age.
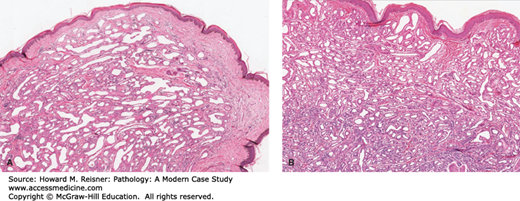
The usual “strawberry” hemangioma (birth mark) seen in infants and children may show rapid growth initially and may be multiple. Grossly, they are bright red to blue in color, are level with the skin surface, and have an intact epithelial surface. Histology shows an unencapsulated aggregate of closely spaced, thin-walled, blood-filled, endothelial-lined capillaries.
Another type of vascular “tumor” is a cavernous hemangioma, a similar collection of larger-caliber vessels usually situated more deeply in tissues or organs (Figure 7-7B).
Vascular ectasias are localized dilatations of preexisting blood vessels usually seen in the skin or mucous membranes that create a focal red lesion.
The nevus flammeus, also known as the “port-wine stain,” is most commonly seen over the face and neck in the distribution of the trigeminal nerve. It may be associated with Sturge–Weber syndrome that includes port-wine stain of the face and venous angiomas of the leptomeninges (covering of the brain) and may cause mental retardation, seizures, and strokes. In patients with nevus flammeus, it is necessary to determine whether there are associated cerebral vascular abnormalities.
A spider telangiectasia is a radial, often pulsatile array of dilated subcutaneous small arterioles usually seen on the face, neck, and chest. They appear to be related to increased systemic estrogen and are often seen in pregnant women and alcoholics with hepatic cirrhosis.
Malformations of dilated capillaries and veins are also seen in autosomal dominant inherited Osler–Weber–Rendu disease. These patients may have these dilated collections of vessels involving their oral cavity, lips, lungs, gastrointestinal tract, or urinary system. As these vascular collections are structurally abnormal, they are at risk for rupture or bleeding (Figure 7-8).
Aortic dissection (also called “dissecting aortic aneurysm”) is a rare but frequently fatal process caused by an intimal–medial tear that allows passage of aortic blood from the true aortic lumen into the medial layer of the aorta creating a false lumen. Aortic dissection has an incidence of 5 to 30 cases per 1 million per year (approximately 2000 cases per year) with a male predominance (2:1) and a peak occurrence in 60- to 70-year-olds. The most common risk factor for an acute aortic dissection in those without an inherited connective tissue disorder (see below) is systemic hypertension (70%–80% of patients). Other risk factors include prior traumatic injury, including iatrogenic, procedure-related injury, aortitis, and medial dysplasia. Cocaine use, which causes a rapid and acute elevation of blood pressure, is also a risk factor and can lead to aortic dissection in younger patients.
CASE 7-2
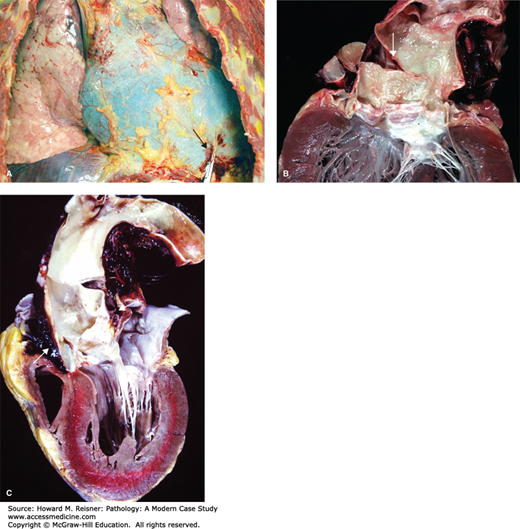
A 67-year-old man, with a history of treated systemic hypertension, suddenly grasped his chest and collapsed while at a local grocery store. Emergency medical services found him unresponsive with a weak pulse. He was rushed to the emergency department where he could not be resuscitated. Autopsy showed a large hemopericardium (blood in the pericardial sac) and an aortic dissection with rupture of the ascending aorta (Figure 7-9).
FIGURE 7-9
Aortic dissection. (A) View of the chest demonstrating hemopericardium. Note extravasated blood at the point of disruption of the pericardium (arrow). From Vincent J. Moylan, Jr., M.S., P.A. (ASCP), Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill. (B) Heart with aorta opened to show the origin of the intimal-medial tear leading to the dissection above the aortic valve (arrow; DeBakey type I; see Figure 7-10). (C) Aorta in cross-section to show blood between the layers of the media (false lumen) (arrow). Figures 7-9b and 7-9c: From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
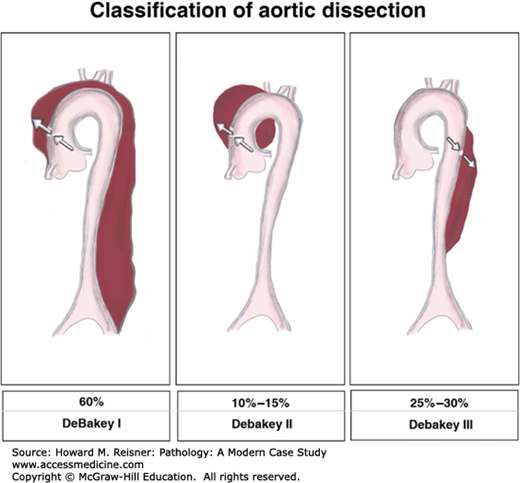
Aortic dissections are classified according to their anatomic location and duration of symptoms. The two most frequently used classification schemes are the DeBakey and the Sanford classifications. The Sanford classification is simplest to understand and is based on the site of the dissection. Sanford type A denotes involvement of the ascending thoracic aorta, regardless of its site of origin. Sanford type B indicates that the ascending aorta is not involved. The DeBakey classification is more complicated, but is most commonly used clinically by surgeons. The most common aortic dissection seen in approximately 54% of cases, the DeBakey type I dissection, indicates that the intimal-medial tear is in the ascending thoracic aorta with the dissection extending distally to the descending thoracic aorta. In DeBakey type II dissections, the intimal-medial tear again is in the ascending thoracic aorta, and the dissection is limited to this region of the aorta. DeBakey type II dissections occur in approximately 20% of patients. In DeBakey type III dissections, the intimal-medial tear is located distally, usually in the distal aortic arch or the proximal descending thoracic aorta. Type III is divided into two subtypes: type IIIa indicates that the dissection, by retrograde extension, involves the ascending thoracic aorta; type IIIb indicates that the dissection is limited to the distal aorta without involvement of the ascending region (Figure 7-10). The determination of the origin of the dissection is crucial for the surgeon who must know where to enter the chest for the aortic repair.
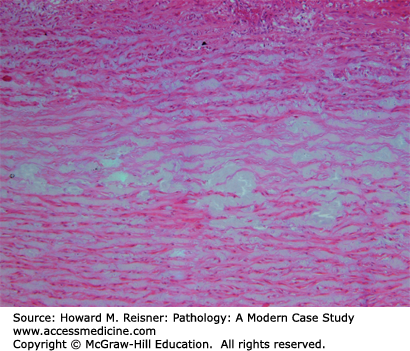
The mechanism of the intimal–medial tear that leads to an acute aortic dissection is presently unknown, although theories abound. The common factor in those with an aortic dissection is acquired or inherited aortic medial weakness (also known as “cystic medial necrosis”). Aortic medial weakness occurs with disruption and interruption of elastic fibers and is a microscopic diagnosis (see below) (Figure 7-11).
FIGURE 7-11
Cystic medial necrosis. The acellular myxoid material (blue) divides and disrupts the elastic fibers of the media. These changes predispose the individual to developing an aortic dissection. From Kemp WL, Burns DK, Brown T. The Big Picture Pathology. New York, NY: McGraw-Hill Lange; 2008. Figure 9-8, page 100.
The pathology of the gross specimen shows a usually horizontal to hockey-stick-shaped intimal-medial tear of the proximal ascending aorta approximately 1 to 3 cm above the aortic valve. Separation between the layers of the aorta with blood clot between the inner and outer medial layers will be seen if the tear is associated with a dissection. Multiple intimal tears and evidence of healing or healed dissections may be seen along the descending thoracic and abdominal aorta as these are most commonly asymptomatic.
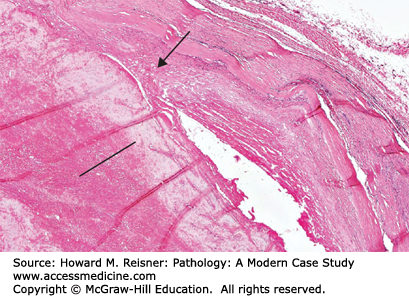
Histology of an acute aortic dissection shows separation between the intima-inner two-thirds of the media and the outer one-third of the media–adventitia with hemorrhage between these layers. In elderly hypertensive patients, the aortic media is usually histologically normal without obvious changes of medial weakness (Figure 7-12).
FIGURE 7-12
Histology of an aortic dissection. An area of medial disruption in the aorta is shown (arrow). The dissecting blood (thrombus) occupies the lower portion of the image (line). (See also Figure 7-9C.) From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
Clinical symptoms are most commonly an abrupt onset of severe chest pain. The location of the pain is usually associated with the location of the dissection: anterior chest pain is associated with dissection involving the ascending aorta (DeBakey type II) and back or interscapular pain is associated with dissection of the distal aortic arch or descending thoracic aorta (DeBakey types I and III). Additional symptoms include syncope, stroke, paraplegia, and sudden death. Often symptoms will vary according to areas of the aorta affected. Patients may present with cough or dysphagia due to compression of the trachea or esophagus by the enlarged, dissected aorta. Other symptoms can result from vascular ischemia, as small arteries are no longer supplied with oxygenated blood from the true lumen.
This disease is important because it is a difficult diagnosis to make. Most commonly diagnosed as an acute myocardial infarct, patients are often sent home when their EKGs and cardiac enzymes are normal. These patients go home and have an increased risk of sudden death due to the significant complications of dissection. There is a high mortality rate if the diagnosis is missed.
The most significant complications include rupture of the false lumen and compression of the true lumen and its arterial branches. Rupture of the false lumen that occurs into the pericardium leads to pericardial tamponade in approximately 90% of proximal dissections. Continued compression of the true lumen and the aortic branches leads to ischemia of the organs deprived of oxygenation. For example, if the carotid artery branches are involved, the brain may be deprived of oxygen leading to cerebral ischemia and stroke (Figure 7-9A).
Disease processes that are associated with aortic dissection include Marfan and Ehlers–Danlos syndromes, congenitally bicuspid aortic valve, aortic coarctation, Turner syndrome, and aortitis. Marfan syndrome, an inherited connective tissue disease, leads to significant aortic medial weakness with large pools of basophilic glycosaminoglycans with significant interruption and disruption of elastic fibers. Patients with Marfan syndrome account for approximately 5% to 10% of all aortic dissections. Approximately one-third of patients with Marfan syndrome develop aortic dissections and must be carefully monitored. Congenitally abnormal aortic valve disease is also associated with a significantly increased risk of aortic dissection. Bicuspid disease has a nine times greater risk of aortic dissection than patients with a normal, tricuspid aortic valve. Patients with a unicuspid aortic valve have a nearly 20-fold increased risk. As awareness has increased over time, this process has been dubbed Bicuspid Aortic Disease (BAD).
Surgical treatment of aortic dissections involves placement of an interposition tube graft at the site of the intimal tear that redirects the blood into the true lumen.
The most significant noninfectious inflammatory diseases of the aorta (aortitis) include Takayasu disease and giant cell aortitis. Multiple systemic connective tissues diseases can also involve the aorta and may have disease-specific features. For example, patients with rheumatoid arthritis (RA) may have rheumatoid nodules in the media of the aorta. Regardless of the underlying etiology, the aorta has limited means of reaction to injury and demonstrates the same gross features of acute and chronic phases regardless of the disease process. The gross features of the acute phase show edema and irregularity of the intimal layer. The gross features of the chronic phase of any aortitis shows marked intimal and adventitial thickening of the vessels with interruption and disruption of the medial elastic fibers and replacement fibrosis. The irregular intimal thickening and the contraction of the scarred media leads to wrinkling of the intima and the classic “tree-bark”-like appearance (Figure 7-13). Like the gross appearance, the histologic appearance of aortitis is not specific; the same type of inflammation, including giant cells, can be seen in noninfectious and infectious aortitis. The incidence, patient features, symptoms, and histology all help differentiate the specific type of aortitis in each case. Also, in the chronic phase, aortitis can lead to aneurysm formation anywhere along the aorta. These aneurysms have an increased risk for rupture.
FIGURE 7-13
Takayasu aortitis. Innominate artery of a 27-year-old woman with Takayasu arteritis with arch vessel involvement. (A) Extensive intimal thickening is seen with attenuation of the media and adventitial fibrosis. (B) Histology revealed degeneration of the media with a dense inflammatory infiltrate, including giant cells (arrow). Image courtesy of Dr. Richard N. Mitchell, Department of Pathology, Brigham and Women’s Hospital. From Gornik HL, Creager MA. Aortic diseases. Aortitis Circ. 2008;117:3039–3051.
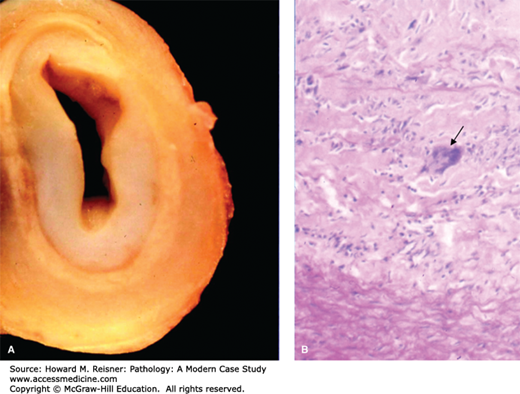
Takayasu disease is a granulomatous inflammation of large- and medium-sized arteries. Multiple synonyms for the disease include pulseless disease, occlusive thromboaortopathy, and young female arteritis. The majority of cases have been seen in Asia and Africa, but cases have been identified worldwide.
Takayasu disease affects women more frequently than men (8:1) most often between 20 and 30 years of age. Approximately two cases per million patients per year are diagnosed in North America and Europe. The disease affects the aorta and the proximal portions of its large branch arteries. Symptoms of the acute phase are similar to all vasculitides and can range from absence of symptoms to fever, weakness, malaise, myalgias, arthralgias, and anorexia. The acute phase can precede the chronic (or occlusive) phase by weeks, months, or years. In the chronic phase, patients may be asymptomatic or may suffer severe systemic complications including stroke, renal artery stenosis, or blindness. Often the patients present with absence of peripheral pulses (96%), bruits (94%), hypertension (74%), and heart failure (28%). The subclavian arteries are involved in approximately 90%, the carotid arteries in 45%, the vertebral arteries in 25%, and the renal arteries in 20%. Coronary arteries are involved in 15% of cases and can lead to myocardial infarct and sudden death.
Multisegmental lesions interrupted by normal artery are changes typical of this disease. Cross-section of occluded arteries has a pale, gray, myxoid gross appearance. In order to make the diagnosis, histology, which must be performed, shows chronic inflammation with perivascular cuffing and medial giant cells and patchy medial necrosis in the acute phase. Histology of the late-stage disease shows scattered smooth muscle cells with rare fibroblasts in the markedly thickened and fibrotic intima and large medial areas with loss of elastic fibers replaced by fibrosis (Figure 7-13).
The exact etiology of the disease is currently unknown. Much research has focused on possible autoimmune mechanisms with identification of susceptibility genes and human leukocyte antigen (HLA) alleles. Familial cases of Takayasu disease do occur.
Syphilitic aortitis is an infectious aortitis, once quite common, accounting for 5% to 10% of all cardiovascular deaths. While it is now less frequent, it is often seen in developing countries and is once again making a come-back in developed countries. Syphilis is caused by infection with the spirochete Treponema pallidum. Syphilitic aortitis shows marked lymphoplasmacytic inflammation involving the vasa vasorum (blood vessels in the adventitia that feed the aortic media). The adventitial vessels are attacked by this inflammatory reaction to the bacteria, causing necrosis of the small arteries leading to fibrosis and scarring with eventual occlusion of the arterioles. The medial layer of the aorta suffers ischemia due to the lack of blood flow from the adventitial vessels, leading to acute necrosis and destruction of elastic fibers with eventual fibrosis and scarring (Figure 7-14). As discussed earlier, the gross and histologic features of syphilitic aortitis are not specific. Therefore, it is necessary to know the patient’s history and serologic status. A patient’s serologic status must be positive in order to diagnose syphilitic aortitis.
Vasculitis is a heterogeneous group of inflammatory processes involving blood vessels of any size in any organ and can give rise to diffuse, nonspecific, constitutional symptoms in patients of all ages. Vasculitides may be classified based on various criteria including infectious versus noninfectious etiology, by size and type of blood vessel involved and specific organ involvement (Figure 7-15).
FIGURE 7-15
Vasculitis versus vessel caliber. Distribution of vessel involvement by large-vessel vasculitis, medium-vessel vasculitis, and small-vessel vasculitis. Note that there is substantial overlap with respect to arterial involvement, and an important concept is that all three major categories of vasculitis can affect any size artery. Small-vessel vasculitis predominantly affects small vessels, but medium arteries and veins may be affected, although immune complex small-vessel vasculitis rarely affects arteries. The diagram depicts (from left to right) aorta, large artery, medium artery, small artery/arteriole, capillary, venule, and vein. Anti-GBM, antiglomerular basement membrane; ANCA, antineutrophil cytoplasmic antibody. From Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11 (Figure 17).
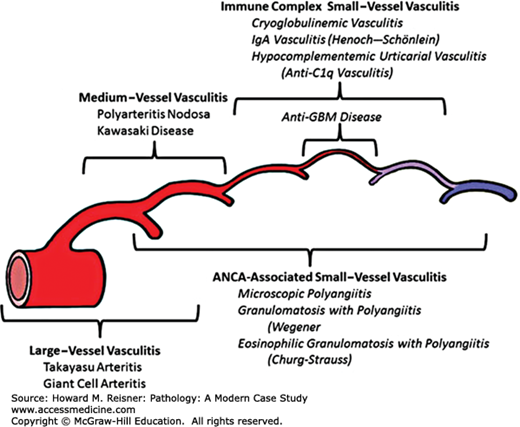
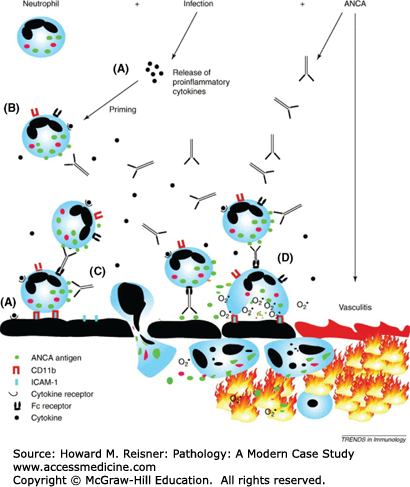
Noninfectious vasculitis is thought to be initiated by immunologic mechanisms including deposition of immune complexes, cell-mediated immune reactions, or direct antibody attack by circulating antibodies. Infectious vasculitis is caused by direct infection by infectious agents and the body’s natural inflammatory reaction to the infection. Inciting agents are largely unknown; however, some types of vasculitis may be associated with viral infections (see Polyarteritis Nodosa section). A distinctive mechanism of immune damage, antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, a form of non-necrotizing vasculitis with little or no deposition of immune complexes, is predominantly associated with small-vessel disease (small-vessel vasculitis [SSV]; Table 7-1). Figure 7-16 provides an overview of the proposed mechanism of parenchymal necrosis and vasculitis in ANCA-mediated vessel damage.
| Vasculitides Nomenclature | |
|---|---|
| Groups | Diseases Included in Group |
| Large-vessel vasculitis (LVV) | Takayasu arteritis (TAK) Giant cell arteritis (GCA) |
| Medium-vessel vasculitis (MVV) | Polyarteritis nodosa (PAN) Kawasaki disease (KD) |
| Small-vessel vasculitis (SVV) | Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AVV) Microscopic polyangiitis (MPA) Granulomatosis with polyangiitis (Wegener) (GPA) Eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome) (EGPA) |
| SVV/immune complex SVV | Antiglomerular basement membrane (anti-GBMD) disease Cryoglobulinemic vasculitis (CV) IgA vasculitis (Henoch–Schönlein) (IgAV) Hypocomplementemic urticarial vasculitis (HUV) (anti-C1q vasculitis) |
| Variable vessel vasculitis (VVV) | Behcet disease (BD) Cogan syndrome (CS) |
| Single-organ vasculitis (SOV) | Cutaneous leukocytoclastic angiitis Cutaneous arteritis Primary central nervous system vasculitis Isolated aortitis Others |
| Vasculitis associated with systemic disease | Lupus vasculitis Rheumatoid vasculitis Sarcoid vasculitis Others |
| Vasculitis associated with probably etiology | Hepatitis C virus-associated cryoglobulinemic vasculitis Hepatitis B virus-associated vasculitis Syphilis-associated aortitis Drug-associated immune complex vasculitis Drug-associated, ANCA-associated vasculitis Cancer-associated vasculitis Others |
FIGURE 7-16
Proposed mechanism of ANCA-mediated neutrophil responses involved in the pathogenesis of ANCA-associated small-vessel vasculitis. (A) Proinflammatory cytokines and chemokines (e.g., tumor necrosis factor α) released as a result of local or systemic infection cause upregulation of endothelial adhesion molecules (e.g., selectins, ICAM-1, and VCAM) and prime the neutrophils. (B) Neutrophil priming causes upregulation of neutrophil adhesion molecules (CD11b) and translocation of the ANCA antigens from their lysosomal compartments to the cell surface. (C) Engagement of the F(ab′)2 portion of ANCA with ANCA antigens on the cell surface and interaction of the Fc part of the antibody with Fc receptors activates the neutrophil, causing increased neutrophil–vessel wall adherence and transmigration. (D) ANCA-mediated neutrophil activation also triggers reactive oxygen radical production and possibly causes neutrophil degranulation, with consequent release of proteolytic enzymes, leading to vasculitis. From Heeringa P, Huugen D, Tervaert JWC. Anti-neutrophil cytoplasmic autoantibodies and leukocyte–endothelial interactions: a sticky connection? Trends Immunol. 2005;26:561–564.
Diagnosis of vasculitis can be quite difficult as the symptoms are nonspecific and often constitutional. Patients may exhibit weakness, fatigue, weight loss, muscle aches (myalgias), and joint pain (arthralgias) during the acute phase. Some of these processes can cause chronic disease with systemic complications. The diagnosis of a specific type of vasculitis is extremely difficult as the symptoms are similar for each type.
Multiple systems of classification of vasculitides have been proposed, but none encompasses both immunopathologic mechanisms and clinicopathologic entities. The recently proposed 2012 International Chapel Hill Consensus Conference system of nomenclature (Table 7-1) has the advantage in linking the names of vasculitides with the caliber of the vessel affected, although other systems remain in use by diagnosticians.
Giant-cell arteritis, a vasculitis that affects all sizes of arteries, is the most common form of systemic vasculitis in adults. It most commonly affects the temporal artery (therefore, is often called temporal arteritis), but may involve any of the cranial arteries and the aorta and its branches. It is rare to make the diagnosis in persons less than 50 years of age (70 years is the average age), with women more commonly affected than men. The incidence increases with age, reaching nearly 1% by 80 years. The age of onset helps differentiate giant cell (temporal) arteritis from vasculitides that can affect the same arteries in younger patients. The self-limited disease is usually benign clinically with symptoms resolving in 6 to 12 months.
Patients most commonly present with throbbing temporal pain, jaw pain, and headaches. The skin over the temporal artery may be swollen, tender, and red. The affected temporal artery may feel cord-like with areas of nodular thickening. Constitutional symptoms may be present including malaise, fever, weight loss, and generalized muscular aches and stiffness of the shoulders and hips (polymyalgia rheumatica). Approximately 50% of patients have visual symptoms, which may progress from transient to permanent blindness in one or both eyes. As the visual symptoms may progress rapidly, they are considered a medical emergency. The disease may also affect the heart, brain, or gastrointestinal system leading to, possibly lethal, specific organ infarction. The diagnosis is made through a 2- to 3-cm biopsy of the temporal artery. Patients’ response to corticosteroids is usually dramatic with symptoms resolving in days.
The disease is segmental, focal, and chronic with granulomatous inflammation that attacks the internal elastic membrane of the artery. The arterial lumen may be reduced to a slit or may be obliterated by thrombosis. As the disease is focal and segmental, multiple levels of the artery are examined microscopically. In a positive biopsy, aggregates of macrophages, lymphocytes, and plasma cells are admixed with variable numbers of eosinophils and neutrophils throughout the media and intima. Giant cells tend to be distributed along the internal elastic lamina, but can vary widely in numbers. Both foreign-body giant cells and Langerhans giant cells are seen. The internal elastic lamina can appear swollen, irregular, and fragmented with foci of necrosis (Figure 7-17). Fragments of the elastica can appear in the cytoplasm of the giant cells. The intima is thickened and the media is fibrotic in later stages. Organized thrombi frequently occlude the arterial lumen, and recanalization can occur. Of note, since the disease is focal and segmental, as many as 50% of biopsies may be negative in patients with otherwise classic symptoms. A negative biopsy does NOT mean that the patient does not have temporal arteritis. If the biopsy is positive, the diagnosis is made definitively.
FIGURE 7-17
Giant cell (temporal) arteritis of a cerebral artery. The temporal artery has a wall that contains a striking inflammatory infiltrate including giant cell formation (arrow). From Kemp WL, Burns DK, Brown T. The Big Picture Pathology. New York, NY: McGraw-Hill Lange; 2008. Figure 9-11, page 103.
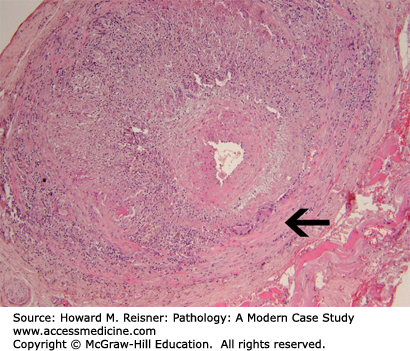
ANCA is negative in giant-cell arteritis. The specific etiology of the disease is unclear, but an association with HLA-DR4 and its occurrence in first-degree relatives support a potential genetic component to its pathogenesis. Morphologic alterations and cellular elements are suggestive of an immunologic reaction. Data suggest that this is a T-cell-dependent disease with activated CD4 T-helper cells. B-cells are absent. Persistent macrophage activation and cytokine production are important features.
Classic polyarteritis nodosa (PAN) is an acute necrotizing vasculitis of medium and small vessels. Like other vasculitides, PAN affects multiple organ systems leading to variable symptoms, although it most commonly affects the skin, joints, peripheral nerves, gastrointestinal tract, and kidney. Of interest, PAN was a rare disease until the 1940s, when there was a marked rise in its incidence. The widespread use of bacterial antisera, animal toxins, and sulfonamides may have been associated with this increase. The diagnosis of PAN currently appears to be decreasing with a current incidence of approximately 3 to 4.5 cases per 100,000 population per year. PAN affects men more frequently than women (1.6–2:1). Typical presentations include fever, night sweats, weight loss, skin ulcerations or tender nodules, and severe muscle and joint pains that develop over weeks to months. It is important to note that the specific patient symptoms will depend on the arterial system involved, resulting in organ dysfunction.
QUICK REVIEW
The American College of Rheumatology has established criteria to help differentiate PAN from other vasculitides. This committee selected 10 disease features associated with PAN; in order to make the diagnosis, at least 3 of the 10 criteria should be present when the diagnosis is made by radiographic or pathologic means. The 10 criteria include:
Weight loss of 4 kg or more
Livedo reticularis (lace-like, purple skin discoloration)
Testicular pain/tenderness
Myalgia or leg weakness/tenderness
Mononeuropathy or polyneuropathy
Diastolic blood pressure >90 mm Hg
Elevated blood urea nitrogen (BUN) or creatinine level unrelated to dehydration or obstruction
Presence of hepatitis B surface antigen or antibody in serum
Arteriogram demonstrating aneurysms or occlusions of visceral arteries
Biopsy of small- or medium-sized artery containing neutrophils
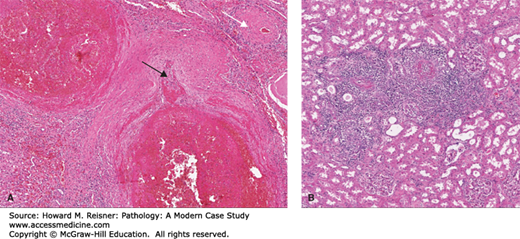
Characteristic lesions of PAN in small- and medium-sized muscular arteries are found in a patchy distribution and may extend into larger arteries. The lesions are usually 1 mm in length and may involve a portion or the entire circumference of the vessel and may present as palpable nodules along the course of medium-sized arteries during autopsy. The lesions affecting medium-sized arteries usually occur at bifurcations or branch points. The histologic appearance depends on the stage of the inflammatory process. Lesions of the acute phase demonstrate fibrinoid necrosis with loss of the muscular media and adjacent tissues with deposition of eosinophilic fibrin material. The fulminant acute inflammatory response is composed of neutrophils, lymphocytes, and plasma cells that vary in their proportions. Eosinophils are often numerous. Lesions in the same artery may vary in age and is a characteristic finding. Due to the vascular injury, luminal thrombosis may occur in the involved segment, leading to ischemia and infarct of the distal organ. Due to the vascular injury and weakening of the vessel wall, aneurysms may occur leading to risk of rupture and significant hemorrhage. Chronic, healed lesions show fibrous proliferation of the intima that may result in significant narrowing of the arterial lumen, again leading to tissue ischemia and potential infarcts (Figure 7-18A).
FIGURE 7-18
(A) Polyarteritis nodosa (PAN). A large nodular lesion demonstrating an aneurysmal outpouching (black arrow) and fibrinoid necrosis. A chronic healed lesion (white arrow) shows intimal proliferation and narrowing of the vessel lumen. (B) Microscopic polyangiitis. Florid inflammation of small arteries/arterioles is present. From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
The exact etiology of PAN is unknown and no animal model is available for study. Interestingly, viral infections including human immunodeficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV) infections have been associated with PAN. A strong association between HBV infection and PAN has been related to immune complex-mediated disease and was once the cause of up to 30% of PAN cases. It is believed that viral replication directly injures the vascular wall causing endothelial dysfunction and production of inflammatory cytokines and adhesion molecules. HBV-associated vasculitis always takes the form of PAN and may occur at any time during the course of the disease. Most commonly it occurs within 6 months of the infection. The widespread use of hepatitis B vaccine has significantly decreased the number of HBV-related PAN that now accounts for approximately 8% of all PAN cases.
Additional associated diseases include other infectious diseases, systemic syndromes, and hematologic malignancies.
Left untreated, the 5-year survival rate of patients suffering from PAN is 13%. Approximately 50% of patients die within the first 3 months of onset. Patients with acute abdominal symptoms have a markedly worse prognosis due to extensive bowel involvement. Their usual postoperative course is complicated by infections and delayed healing. Central nervous system involvement has a worse prognosis than does peripheral nerve involvement. Patients with cutaneous PAN without systemic involvement have a benign prognosis, although the disease tends to relapse. Death is most commonly due to uncontrolled vasculitis, infectious complications due to immunosuppression, and vascular complications including myocardial infarction (MI) and stroke. Corticosteroid therapy improves the 5-year survival rate to nearly 50% to 60% to greater than 80% when combined with other immunosuppressants.
Microscopic polyangiitis (MPA; formerly known as microscopic polyarteritis) is an ANCA-associated systemic vasculitis with some features similar to PAN with involvement of small arteries and arterioles and the microcirculation (renal glomeruli and pulmonary capillaries) (Figure 7-18B).
Kawasaki disease (KD), also known as mucocutaneous lymph node syndrome, is a pediatric inflammatory illness of unknown etiology. This disease process is the leading cause of acquired heart disease in childhood in North America and Japan. The disease is endemic in Japan with an incidence of 20 to 100 cases/100,000 population <5 years of age. In North America, the incidence is 4 to 15 cases/100,000 population. The disease most commonly affects (in order of frequency) Asians, African-Americans, Whites, and Native Americans with a slight male predominance (1.5:1). Eighty percent of affected children are less than 4 years of age. Clinical symptoms include an abrupt high fever that lasts longer than 5 days, conjunctival and oral erythema with erosions (“strawberry” tongue), edema of the hands and feet, erythema of the palms of the hands and soles of the feet, a diffuse skin rash with desquamation, and enlargement of cervical lymph nodes. The disease, similar to other pediatric viral illnesses, is usually self-limited, even without treatment. Although it appears similar to other viral illnesses, no specific infectious cause has been identified. Infections with parvovirus B 19 or with New Haven coronavirus have been implicated in some cases. Various bacterial infections (including Staphylococcus, Streptococcus, and Chlamydia) have been implicated in occasional cases. The common theme in such cases appears to be production of super-antigens that bind to major T-cell receptors resulting in massively activated immune responses in an antigen-nonspecific manner. Autoantibodies to endothelial and smooth muscle cells have been identified in other patients.
CASE 7-3
A previously healthy, 17-year-old was playing basketball with his friends. He told them he needed a break and went into the house. He didn’t return and his friends found him dead on the floor of the kitchen.
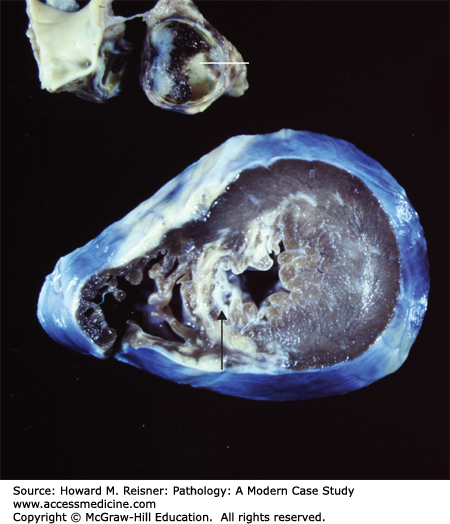
Pathology: At autopsy, his heart was markedly enlarged with dilated, aneurysmal coronary arteries. Cross sections showed the coronary arteries (Figure 7-19 top) had a thickened wall with intimal fibrosis and thrombotic occlusion of the anterior descending and right coronary arteries. Histology showed a healed arteritis of the aneurysmal coronary arteries. Both histology and gross examination (Figure 7-19 bottom) revealed evidence of ongoing ischemic injury and scarring of the myocardium.
This young man’s cause of death: sudden cardiac death due to myocardial ischemia following occlusion of giant coronary artery aneurysms due to healed Kawasaki disease.
FIGURE 7-19
Kawasaki disease (radiograph). A thrombosed large aneurysm of the left descending coronary artery (line) led to the sudden death of this 15-year-old individual. Marked fibrosis of the left ventricular septum (arrow) and wall is related to long-standing myocardial ischemia associated with the vasculitis. From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
The acute phase of the disease causes an acute necrotizing vasculitis similar to PAN that specifically affects the epicardial coronary arteries. Approximately 70% of patients have coronary artery involvement resulting in death in 1% to 2%.
The most important cardiovascular sequelae occur in approximately 20% of patients, ranging from asymptomatic vasculitis to giant coronary artery aneurysms (Figure 7-19). Giant coronary artery aneurysms (7–8 mm) occur in 25% of children and are usually multiple. Significantly, the aneurysms may thrombose leading to myocardial ischemia and infarction.
During the acute phase, the histology is similar to that of PAN, with patchy vascular fibrinoid necrosis with varying collections of neutrophils, lymphocytes, plasma cells, and eosinophils. Of note, the disease is associated with activation of T-cell lymphocytes and macrophages that results in elevated levels of inflammatory cytokines. The resultant, healed aneurysms show destruction and absence of the elastic laminae, medial thinning, and fibrointimal proliferation and organized mural thrombi.
Persistence rate of aneurysms is 72% at 1 year and 40% at 5 years after disease occurrence. Approximately 55% of patients show regression of aneurysms over a significant period (14 years). Regression is inversely related to severity of the initial aneurysm and male gender. Treatment with high-dose aspirin and intravenous gamma-globulin is positively related to aneurysm regression.
After treatment, the long-term rate of an acute myocardial infarct is approximately 1% with a mortality rate of 1%. Patients dying of ischemic sequelae of KD show occlusion of one or more coronary arteries in 75% of cases.
Granulomatosis with polyangiitis (previously known as Wegener granulomatosis) is a systemic necrotizing vasculitis of unknown etiology that manifests as granulomatous lesions of the respiratory tract including the nose, sinuses, and lungs and as a renal glomerular disease. Men are more commonly affected in their fifth and sixth decades. Studies have demonstrated ANCAs in the blood of approximately 90% of patients; 75% of these are positive for cytoplasmic-ANCA (c-ANCA more currently identified as proteinase-3 (PRTN-3)). It has been suggested that c-ANCAs may activate circulating neutrophils to attack the blood vessels.
Patients usually present with respiratory symptoms including pneumonitis and sinusitis. Chronic sinusitis and nasopharyngeal mucosal ulcers are frequent. The lung is ultimately involved in over 90% of patients. Radiologically, multiple pulmonary infiltrates are prominent and may be cavitary. Hematuria and proteinuria are common due to focal necrotizing glomerulonephritis. Progression to crescentic glomerulonephritis can lead to renal failure. Rash, muscular pains, joint involvement, and neurologic symptoms can occur. Untreated, the disease leads to an 80% mortality rate within 1 year with a mean survival of 5 to 6 months.
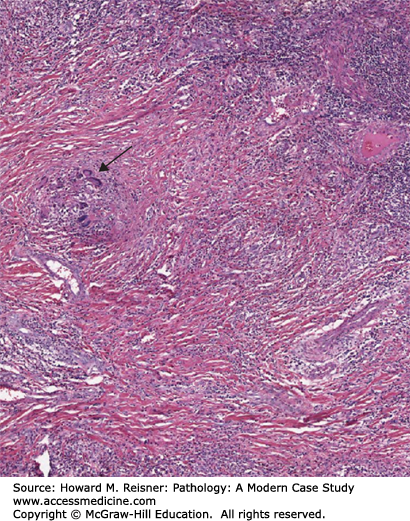
Gross pathologic lesions of polyangiitis with granulomatosis show parenchymal necrosis with a geographic pattern and individual lung lesions as large as 5 cm in diameter which may cavitate, giving an appearance similar to those seen in tuberculosis. The vasculitis involves small arteries and veins most frequently in the lung, kidney, and spleen. Histologically, the vasculitis is often characterized primarily by chronic inflammation, but acute inflammation, necrotizing and non-necrotizing granulomatous inflammation, and fibrinoid necrosis are often present. Later in the disease, medial thickening and intimal proliferation are common and can lead to narrowing or obliteration of the vessel lumen (Figure 7-20) resulting in tissue ischemia.
FIGURE 7-20
Granulomatosis with polyangiitis (Wegener granulomatosis). Disease in lung with granulomatous inflammation indicated by an arrow. There is extensive parenchymal necrosis and vasculitis throughout the image. From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
Treatment with cyclophosphamide produces complete disease remission and substantial disease-free intervals in most patients. Of note, antimicrobial sulfa drugs significantly reduce the incidence of relapses.
Eosinophilic granulomatosis with polyangiitis is a systemic vasculitis with prominent eosinophilic inflammation that affects young persons with asthma or allergies. Churg–Strauss syndrome (CSS) is a rare disease. There is no gender predilection. Several exogenous trigger factors for disease onset or flares have been suspected in some studies and include vaccinations, desensitizations, and medications
Its most typical presentation consists of the appearance of vasculitis manifestations including fever, rash, and central nervous system symptoms in a patient with known allergic rhinitis, nasal and sinus polyposis, and asthma. The combination of blood eosinophilia and inflammatory symptoms is highly suggestive of the diagnosis. Thirty to forty percent of patients with CSS are positive for perinuclear-ANCA with anti-myeloperoxidase specificity.
In addition to the asthma with airflow obstruction, patchy and transient alveolar infiltrates with pleural inflammation can occur. Allergic rhinitis, sinusitis, and/or nasal polyps can be seen in as many as 60% to 80% of patients. Skin lesions including purpura, nodules, and hives occur in approximately 50% of patients. Peripheral neuropathy affects about 50% to 80% of patients and can be severe. Renal manifestations occur in up to 25% of cases and range from isolated urinary abnormalities to rapidly progressive renal failure. Patients with CSS must be evaluated for cardiac involvement, as such carries a poor prognosis.
Pathology shows widespread necrotizing lesions of medium- and small-sized arteries, arterioles, and veins primarily of the lungs, kidney, spleen, heart, liver, and central nervous system. The lesions are granulomatous with a prominent eosinophilic infiltrate in and around the vessels, resulting in fibrinoid necrosis, thrombosis, and aneurysm formation similar to PAN. The resultant occluded vessels lead to distal organ ischemia and possible infarct (Figure 7-21).
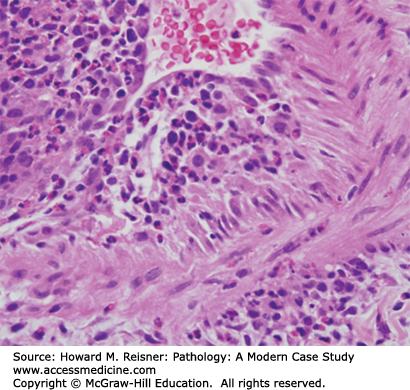
It is important to distinguish CSS from other diseases with eosinophilia including parasitic and fungal infections, granulomatosis with polyangiitis, eosinophilic pneumonia, and hypersensitivity vasculitis.
Untreated patients have a poor prognosis. Corticosteroid therapy is almost always successful in leading to remission.
Hypersensitivity vasculitis refers to a broad group of inflammatory vascular lesions thought to represent a reaction to foreign materials. The skin is the most commonly affected organ in primary hypersensitivity reactions, although any organ may be involved. In the case of lesions confined predominately to the skin, a definitive diagnosis rests on histopathologic examination of the affected tissues. A tissue diagnosis is easily accomplished with cutaneous involvement. Cutaneous vasculitis may occur following administration of many drugs, various bacterial infections (streptococcal and staphylococcal), viral hepatitis, tuberculosis, and bacterial endocarditis. The disease typically presents as palpable purpura, predominately on the lower extremities. Cutaneous vasculitis is usually self-limited, even without treatment.
CASE 7-4
A six-year-old boy was brought to his pediatrician with symptoms consistent with group A beta-hemolytic streptococcal pharyngitis and was prescribed a course of amoxicillin. After several days of treatment, the boy complained of severe itching on both forearms, and an extensive rash was noted by his mother. The mother thought the rash was due to “hives” but the boy became increasingly uncomfortable and was returned to the pediatrician. On examination, the pediatrician noted a palpable purpuric rash consisting of multiple 2-mm lesions on his forearms and thighs. He complained of joint pain. A urinalysis was normal. The rash persisted. Concerned over the possibility of severe disease, the pediatrician performed a skin biopsy in an affected region.
The skin biopsy (Figure 7-22) was interpreted as hypersensitivity vasculitis likely related to drug sensitivity. Antibiotics had been discontinued and the rash resolved spontaneously.
Pathology: The lesion was described as demonstrating vascular and perivascular infiltrates predominantly of neutrophils with leukocytoclasia.
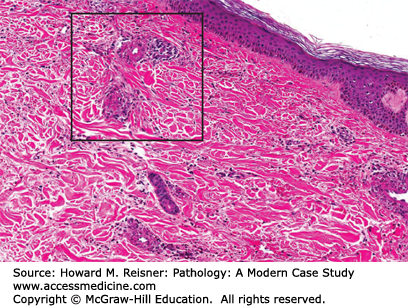
The most common histologic pattern is a neutrophilic infiltrate of postcapillary venules associated with leukocytoclasia (destruction of white blood cells with karyorrhexis) and evidence of vascular damage including EC swelling, fibrinoid necrosis, and red blood cell extravasation. The vascular lesions of hypersensitivity vasculitis are all at the same stage of development at any given time (Figure 7-22).
Systemic hypersensitivity angiitis may be an isolated entity or may be a feature of other disease processes including collagen vascular diseases and a variety of malignancies. Patients with systemic hypersensitivity angiitis may also have skin manifestations. Small-vessel angiitis may cause microinfarcts of internal organs, which do not usually cause major organ dysfunction.
Subtypes of hypersensitivity vasculitis are all characterized by an immunologically mediated, small-vessel angiitis. They may be distinguished by differing clinical presentations, specific laboratory findings, and/or the presence of an underlying systemic disorder.
Serum sickness is a clinical syndrome resulting from injection of heterologous serum or serum proteins. Classic serum sickness is infrequent today, as heterologous serum is rarely used. However, reactions to nonprotein-containing drugs are common.
Henoch–Schönlein purpura, also known as anaphylactoid purpura, is a systemic small-vessel vasculitis. It appears clinically as a syndrome of nonthrombocytopenic purpura, arthritis, gastrointestinal bleeding, and renal disease. This disease is caused by deposition of immune complexes due to viral or streptococcal infections, medication administration, and ingestion of certain foods. Unlike hypersensitivity vasculitis, IgA is principally detected as part of the circulating immune complex and/or immune deposits within vessels and renal glomeruli (see Chapter 12).
Vasculitis associated with mixed cryoglobulinemia. Diagnosis of this condition is made by the demonstration of a cryoglobulinemia and a small-vessel vasculitis.
Vasculitis associated with other systemic diseases. Necrotizing small-vessel vasculitis has been associated with a number of collagen vascular diseases including systemic lupus erythematosus (SLE), RA, systemic sclerosis, and Sjögren syndrome. Similar small-vessel vasculitides have also been associated with various neoplastic conditions including myeloproliferative disorders, hairy cell leukemia, lymphoproliferative disorders, multiple myeloma, and a variety of carcinomas.
As its name suggests, thromboangiitis obliterans (TAO) is an occlusive disease of arteries in the arms and legs almost exclusively seen in young and middle-aged men who smoke heavily. Previously, the disease was thought to only occur in men; however, increasing numbers of affected women are being reported. While most patients are heavy smokers, the disease has also been reported in cigar smokers and users of smokeless tobacco, demonstrating the significant association between tobacco and the disease. The disease has not been substantiated in patients without some form of tobacco use. TAO is more common in the Mediterranean areas, the Middle East, and Asia. Interestingly, while tobacco is central to the initiation and continuance of the disease, EC dysfunction has been found in arteries not yet clinically involved with the disease. Elevated levels of antiendothelial cell antibodies have also been found in these patients.
Typically, TAO occurs in smokers with the onset of symptoms before the age of 40 to 45 years. The disease usually begins with involvement of the distal small arteries and veins. The patients usually complain of claudication (literally, to limp; used to describe a cramp-like pain most commonly due to decreased blood flow or ischemia) of the feet and legs and occasionally of the arms and hands. As it progresses, the disease may involve more proximal vessels with development of ischemic ulcerations of the distal portions of the toes or fingers. Seventy-six percent of patients had ischemic ulcerations at the time of presentation. Two or more limbs are usually involved in the disease process. No specific laboratory tests are diagnostic of Buerger disease. However, it is important to exclude other diseases that might mimic TAO such as hypercoagulable states, other systemic diseases and illicit drug use. Cocaine and amphetamines ingestion can mimic TAO and show nearly identical arteriographic findings. Therefore, in patients with manifestations of Buerger disease who do not use tobacco, a toxicology screen can be helpful. Angiographic features show multiple segmental occlusions of small- and medium-sized vessels with collateralization around the obstructions (corkscrew collaterals).
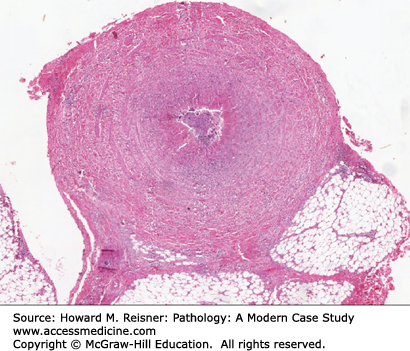
The histopathology of the acute phase of the disease is the most diagnostic feature of the disease (Figure 7-23). Biopsy of an acute superficial thrombophlebitis is most likely to show the characteristic lesion of the acute phase. Acute inflammation involves all layers of the vessel wall and is associated with an occlusive, cellular thrombosis. Degenerating neutrophils are at the periphery of the thrombus, and multinucleated giant cells may be seen. The subacute lesion shows organization of the occlusive thrombus. Decreased numbers of inflammatory cells are seen in the vessel wall in the subacute phase. The chronic phase (or end stage) shows the vessel lumen occluded by an organized thrombus. Recanalization of the thrombus may be prominent. Of note, the normal architecture and the elastic laminae of the vessel wall remain intact and allow differentiation from other vasculitides.
FIGURE 7-23
Thromboangiitis obliterans (Buerger disease). Biopsy of an area of acute superficial thrombophlebitis demonstrates multilayer inflammation of the vessel wall and an occlusive cellular thrombus surrounded by cellular debris. From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
Therapy consists of the complete discontinuation of cigarette smoking and the use of any form of tobacco. Complete abstinence is the only way to halt the progression of TAO and to avoid future complications, including amputations. All other forms of treatment are palliative. The risk of amputation in previous smokers is eliminated 8 years after smoking cessation.
Behçet disease is a chronic and multisystem inflammatory disorder with a remarkable ability to involve blood vessels of nearly all types and sizes and involving both veins and arteries. Manifestations may occur at many sites throughout the body because of the diversity of the blood vessels affected, although the disease has a predilection for certain organs and tissues including the eye, mouth, skin, lungs, joints, brain, gastrointestinal tract, and genitals. The disease is most commonly seen in populations from Japan and China to the Mediterranean Sea and develops in persons aged 20 to 30 years.
Patients may develop anterior and/or posterior uveitis (inflammation of the middle, pigmented vascular structures of the eye including the iris, ciliary body and choroid) of the eye. Anterior uveitis causes blurry vision, pain, light sensitivity, tearing, and redness of the eye. Posterior uveitis can be more dangerous with few physical symptoms and can lead to blindness due to damage to the retina.
Painful aphthous ulcers of the mouth are similar to mouth ulcers in the general population; however, they are more numerous, more frequent, larger, and more painful in Behçet disease. These ulcers can be found on the lips, tongue, and the inside of the cheeks and may be single or appear in clusters.
Genital ulcers develop on the scrotum or vulva and are similar to the mouth ulcers, but frequently involve deeper tissues.
Involvement of the central nervous system is one of the most dangerous manifestations of Behçet disease. The disease tends to involve the white matter of the cerebrum and brainstem. Symptoms may include headache, confusion, strokes, personality changes, and dementia. It may also present as “aseptic” meningitis.
Pustular skin lesions can occur anywhere on the skin. Red, tender nodules that occur on the legs and ankles (erythema nodosa) may also appear on the face, neck, or arms. Unlike erythema nodosa associated with other diseases, the skin lesions associated with Behçet disease tend to ulcerate. Aneurysms in the lung may rupture leading to massive lung hemorrhage. Joints may develop arthritis. Ulcerations may occur anywhere in the gastrointestinal tract from the mouth to the anus, most commonly in the terminal ileum and cecum.
Behçet disease is one of the only forms of vasculitis that has a genetic predisposition. The presence of HLA-B51 is a risk factor for the disease; however, only approximately 5% of cases are familial. Therefore, it is believed that other factors play a role in developing the disease in the presence of HLA-B51. Possibilities include infections (Streptococci) and environmental exposures.
The underlying pathologic lesion is a small-vessel vasculitis that predominately affects venules. Biopsies of mucocutaneous lesions show a perivascular infiltration of lymphocytes, plasma cells, and monocytes affecting arterioles and venules. Frank necrosis of small vessels is unusual. Cutaneous vasculitis appears mainly as a venulitis or phlebitis.
Treatment for disease confined to mucocutaneous regions is topical steroids and medications such as colchicine. Corticosteroids may be required for disease exacerbations with some patients requiring low-dose steroids to keep the disease under control. Serious disease with end-organ involvement requires high-dose corticosteroids and other immunosuppressive agents.
Acute radiation vasculitis injury shows EC damage, ballooning degeneration of the intimal cells, and necrosis of medial smooth muscle with or without thrombosis of the small arteries and arterioles. The chronic phase shows fibrous intimal proliferation that may lead to significant luminal narrowing. Radiation can lead to significant luminal narrowing of the coronary arteries with resultant myocardial ischemia after chest radiation.
HYPERTENSIVE VASCULAR DISEASE
Systemic hypertension is one of the most important modifiable risk factors for atherosclerotic coronary heart disease, stroke, congestive heart failure, and end-stage kidney disease. Prevention and treatment of hypertension can lead to a significant decrease in morbidity and mortality from the leading causes of death in North America. The effects on end organs increase as the blood pressure increases.
Specific numbers for the systolic and diastolic blood pressure levels are based on epidemiologic studies that relate blood pressure levels to risks for adverse outcomes and on evidence from clinical trials that show a reduced risk of adverse outcomes when hypertension is controlled. The question of what is adequate control has, until recently, been based on the recommendations from the Seventh Report of the Joint National Commission of Prevention, Detection, Evaluation and Treatment of High Blood Pressure (JNC VII) released in 2003 which classify blood pressure for adults 18 years or older:
Normal—Systolic <120 mm Hg, diastolic <80 mm Hg
Prehypertension—Systolic 120 to 139 mm Hg, diastolic 80 to 90 mm Hg
Stage I—Systolic 140 to 159 mm Hg, diastolic 90 to 99 mm Hg
Stage II—Systolic ≥160 mm Hg, diastolic ≥100 mm Hg
These blood pressure classifications are based on an average of 2 or more readings taken at each of two or more visits after an initial screening. Prehypertension is a new category designated in the JNC VII and is important as it emphasizes that these patients are at risk to progress to hypertension.
These recommendations have been recently reevaluated in the 2014 Evidence-Based Guideline for the Management of High Blood Pressure in Adults Report From the Panel Members Appointed to the Eighth Joint National Committee (JNC 8) (Table 7-2). These guidelines establish thresholds for pharmacologic management of persons based on age and clinical status rather than establishing categories of hypertension. For example, treatment thresholds for nondiabetic, nonrenal disease patients have been defined as greater than systolic 140, diastolic 90 for persons between 18 but less than 60 years of age and systolic 150, diastolic 90 for persons of 60 years old.
| All adults aged ≥ 18 years with hypertension | ||
| Implement lifestyle intervention | ||
Set blood pressure goals Initiate blood pressure lowering medication | ||
Age ≥ 60 yr with no diabetes or CKD Blood pressure goal SBP < 150 mm Hg DBP < 90 mm Hg | Age < 60 yr or all ages with diabetes but no CKD Blood pressure goal SBP < 140 mm Hg DBP < 90 mm Hg | All ages with CKD with/without diabetes Blood pressure goal SBP < 140 mm Hg DBP < 90 mm Hg |
Non-Black: Initiate thiazide-type diuretic or ACEI or ARB or CCB alone or in combination Black: Initiate thiazide-type diuretic or CCB alone or in combination | All races: Initiate ACEI or ARB, alone or in combination with another drug class | |
Blood pressure is determined by cardiac output and blood volume (pressure = flow × resistance). Blood volume is dependent on sodium and water balance. Blood vessel resistance, regulated primarily at the level of the arterioles, is dependent on hormonal and neural systems. Hormonal components such as the renin–angiotensin, kinin, and prostaglandin systems play important roles in vascular tone. Other factors, including hypoxia and acidosis, can cause significant alterations in vascular resistance.
Clinically, systemic hypertension is divided into two categories: essential (idiopathic) or secondary. Essential hypertension accounts for about 95% of the 50 million Americans with hypertension. Secondary hypertension related to a definable clinical cause makes up less than 5% of cases. Patients presenting with new-onset hypertension should undergo a complete work-up for causes of secondary hypertension, as some of these causes are immediately treatable. Secondary hypertension can be caused by renovascular hypertension (renal artery stenosis, fibromuscular dysplasia), endocrine diseases (including pheochromocytoma, exogenous steroid administration), neurogenic causes, drugs, and toxins.
After ruling out causes of secondary hypertension, the remaining 95% of hypertensive cases are considered essential or idiopathic. The pathogenesis of the disease is multifactorial and its etiology has been difficult to completely determine due to the many factors involved in blood pressure modulation, including the vasculature, the central and sympathetic nervous systems, the kidney and many hormone regulators involved, and the effects of diet and medications. Currently, it is believed that multiple factors contribute to the development of hypertension, including an inherited predisposition, excessive dietary salt intake, and increased adrenergic tone.
While epidemiologic and family aggregation studies have definitely established that there is a significant inheritable component for blood pressure, how genetics plays a role has not yet been determined. While the study of rare genetic disorders affecting blood pressure has led to the identification of uncommon causes of hypertension, these have not contributed to the determination of specific genes that may be responsible for hypertension in the general population. Environmental factors are believed to alter the genetic components in many familial cases. The heavy intake of salt has been one factor that has been shown to significantly increase the risk of developing hypertension in genetically predisposed individuals.
The kidney has a central role in maintenance of blood pressure by its ability to regulate sodium and water. Many researchers have shown that the kidney also plays a major role in the development of high blood pressure. The development of hypertension has been demonstrated in the kidney through loss of sodium excretory pathways leading to increased intra-arterial pressures, cross-transplantation studies where the hypertension was transmitted to the recipient by the donor kidney and the major role played by the renin-angiotensin system. One study demonstrated the importance of the nervous system in hypertension by their ability to lower blood pressure in patients with refractory hypertension by denervation of the kidney.
Other studies have revealed the importance of changes in vascular tone as a crucial component of hypertension. The elevated peripheral resistance demonstrated by hypertensive patients, mediated by hormonal regulators, may be altered by targeting specific genes and their signaling pathways. The possibility of hypertension being an autoimmune disease process has also been proposed. Of interest, using this theory, some researchers are attempting to develop a vaccine for hypertension.
Systemic hypertension, associated with increased atherogenesis, also causes degenerative changes in large- and medium-sized blood vessels. These degenerative changes cause weakness of the vascular wall that can be responsible for rupture of the aorta (aortic dissection) or cerebral vasculature (intracerebral hemorrhage [see above]). Small arteries show hyaline arteriolosclerosis, a pink, homogeneous, hyaline-like thickening of the arterial wall. The arteriolar lumen is most commonly narrowed, leading to decreased flow of oxygenated blood to distal organ structures. The hyalinized appearance of the arterioles is due to leakage of plasma components across the vascular endothelium (due to damage by the increased systemic pressures) and increased production of extracellular matrix by smooth muscle cells. Hyaline arteriolosclerosis is one of the major components of benign nephrosclerosis (see Hypertensive Arterionephrosclerosis section in Chapter 12).
Like the heart lung and kidneys, systemic hypertension causes significant structural changes in the vasculature of the brain. Both systolic and diastolic pressures are important predictors for primary and recurrent stokes. The specific pathological changes are described as both lipohyalinosis (deposition of lipid and hyaline material into the arterial wall) and/or fibrinoid necrosis. The chronic changes result in thickening of the arterioles with deposition of type IV collagen into the arteriolar wall, leading to loss of normal contractile function. Cerebral hypertension may result in spontaneous intracerebral hemorrhage. Aneurysm-like lesions found at sites of the rupture are a likely precursor lesion leading to the acute intraparenchymal hemorrhages. These miliary aneurysm-like lesions are more commonly detected in hypertensive individuals.
Intracerebral hemorrhage in hypertensive patients is a life-threatening disease that carries significant morbidity and mortality. In these patients, the alterations in the arterioles allow blood to extravasate into surrounding brain parenchyma causing destruction of adjacent tissues and possible rupture of the hemorrhage into ventricles or the subarachnoid and subdural spaces (see Case XXX and Chapter 21 “Brain Hemorrhage”).
Both acute and chronic hypertensive changes may manifest in the eye and range from acute changes of malignant hypertension (see below) to chronic changes from long-term hypertension. As is the case with the cerebral vasculature, retinal vasculature becomes thickened in the setting of chronic hypertension. The normal light reflex of the retinal arterial system is formed by the reflection from the interface between the column of blood and the vessel wall. In hypertension, the progression of hyalinization and thickening causes the light reflex to become more diffuse and the retinal arterioles appear red-brown (copper wire). As the sclerosis advances, the increased density of the retinal arterioles seen on ophthalmoscopy is referred to as sheathing of the vessels. When the sheathing encircles the entire wall, it produces an appearance of a silver wire. Focal narrowing of arterioles due to arteriolosclerosis can impede forward circulation resulting in dilated veins peripheral to the arteriolar/venous crossing. This is known as arteriovenous nicking (Gunn sign). Additional retinal changes associated with chronic hypertension include retinal hemorrhages, loss of nerve fiber layers, increased vascular tortuosity, and vascular remodeling due to capillary nonperfusion (microaneurysms and shunt vessels).
CASE 7-5
A 63-year-old woman with a history of poorly controlled hypertension was brought to the emergency department after collapsing at home. She had been complaining of severe headaches for 2 days. A CT of the head showed a large acute intracerebral hemorrhage with extension into the ventricles (Figure 7-24a). Her neurologic status continued to decline and she was declared brain dead. At autopsy, her heart and kidneys showed chronic hypertensive changes. Examination of the brain showed diffuse brain swelling with massive intracerebral hemorrhage involving the left thalamus and basal ganglia (Figure 7-24b) with extension of the hemorrhage into the ventricular system. Microscopic examination showed widespread acute intraparenchymal hemorrhage. Surrounding brain tissueshowed thickened, sclerotic small arteries with perivascular hemosiderin-laden macrophages (Figure 7-24c). (Case provided courtesy of Drs. Diane Armao and Thomas Bouldin Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.)
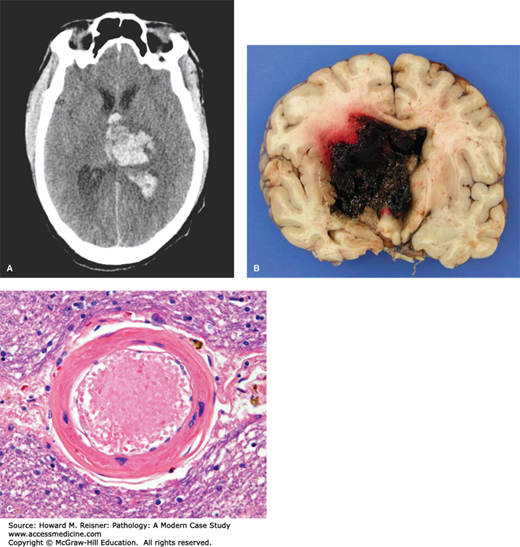
Malignant hypertension, a medical emergency, is the rapid and severe elevation of blood pressure that may lead to end-organ damage of the heart, kidneys, and/or brain. Most commonly, the systolic blood pressure is elevated to 180 mm Hg or higher. Malignant hypertension is much less frequent than idiopathic systemic hypertension, occurring in only a small percentage of patients with elevated blood pressures. Those found to be at risk of developing malignant hypertension are African-American men or someone of a lower socioeconomic status. Poor access to health care also increases the risk. Malignant hypertension may develop spontaneously in a previously healthy individual or as a complication in approximately 1% of previously hypertensive patients. Other causes include any form of secondary hypertension, pregnancy, cocaine, monoamine oxidase inhibitors (MAOIs), oral contraceptives, withdrawal of alcohol, β-blockers, or α-stimulants.
The precise etiology of malignant hypertension is unclear, but the initial event appears to be vascular damage to the kidneys. In the setting of chronic hypertension, it may result from end-stage changes to the arterioles or may be in the setting of arteritis. Injury to the arterioles leads to increased permeability of the small vessels to plasma proteins (including fibrinogen), platelet deposition, and endothelial cell injury. The plasma proteins and fibrin are deposited into the wall of the arteriole, giving the vessels an eosinophilic, homogeneous, and granular appearance (fibrinoid necrosis, Figure 7-25a). The arterioles may develop occlusive intraluminal thrombosis leading to renal ischemia. Interlobar arteries and larger arterioles show a different response with proliferation of intimal (believed to be smooth muscle) cells that produce a concentric arrangement with an onion-skin appearance (Figure 7-25b). This hyperplastic arteriolosclerosis causes severe narrowing of the arterial and arteriolar lumens and further ischemic changes of the kidneys.
FIGURE 7-25
Malignant Hypertension in Renal Vasculature. (A) Fibrinoid necrosis N deposited amorphous eosinophilic material in vessel wall. (B) “Onion-skin” changes demonstrating proliferation of intimal (smooth muscle) cells. From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
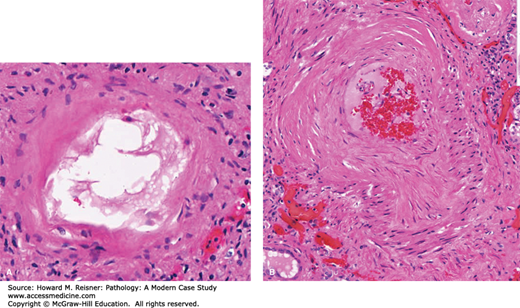
Early symptoms of malignant hypertension depend primarily on the organ system most severely affected initially and can include chest pain, heart failure, headache, and visual disturbances, The most severe symptomatology of malignant hypertension is characterized by diastolic blood pressures >120 mm Hg, papilledema, encephalopathy cardiovascular abnormalities, and renal failure. If untreated, malignant hypertension causes significant morbidity and mortality. Often fatal complications include aortic dissection, myocardial ischemia, heart failure, intracerebral hemorrhage, and acute renal failure.
Therapy is to initially reduce the mean arterial blood pressure by approximately 25% over the first 24–48 hours as a rapid reduction may lead to organ hypoperfusion and organ ischemia. Prior to the time of effective treatment, the life expectancy of patients with malignant hypertension was less than 2 years. With current therapy and renal dialysis as a bridge to returning renal function, the survival rate at 1 year is >90% and at 5 years is approximately 80%.
The pulmonary vascular system is one of low resistance with normal arterial pressures approximately one-tenth of systemic (at rest <20 mm Hg). Pulmonary hypertension, defined as mean pulmonary arterial pressures ≥25 mm Hg, presents equally in men and women of all age groups. The mean age at diagnosis is in the 3rd and 4th decades. The current WHO classification system divides pulmonary hypertension (PH) into 5 groups based on the etiology of the condition:
Group 1: Patients with pulmonary arterial hypertension including idiopathic pulmonary arterial hypertension, heritable idiopathic pulmonary arterial hypertension, and PH that is localized to small pulmonary muscular arteries including connective tissue diseases, congenital heart disease, pulmonary venoocclusive disease, HIV infection, persistent pulmonary hypertension of the newborn, and parasitic infections.
Group 2: Pulmonary hypertension due to left heart failure, including systolic or diastolic dysfunction or valvular heart disease.
Group 3: Pulmonary hypertension due to lung disease or hypoxemia including chronic obstructive disease, interstitial lung disease, obstructive sleep apnea, and hypoventilation syndrome.
Group 4: Chronic thromboembolic pulmonary hypertension. These patients have pulmonary hypertension due to vascular obstruction by recurrent thromboemboli.
Group 5: Pulmonary hypertension due to unknown or multifactorial mechanisms.
Pathology: The underlying pathology of pulmonary artery hypertension is dependent on its etiology. In chronic thrombotic/embolic disease, pulmonary arteries and arterioles will show eccentric mounds of acute, organizing and organized thrombus along intimal surfaces. In patients with left heart failure, the major changes of intimal fibrosis will be identified in pulmonary veins.
The morphology of PAH seen in patients with unoperated congenital heart disease includes six grades of structural changes in the pulmonary arteries. Grades 1–3 show progressive medial thickening with adventitial fibrosis in pulmonary arterioles and muscular pulmonary arteries. Grade 3 shows progressive intimal proliferation and fibrosis in smaller pulmonary arteries and arterioles with extension into medium-sized arteries. The characteristic feature of grade 3 pulmonary vascular disease is the widespread occlusion of muscular pulmonary arteries and arterioles by acellular fibrous tissue admixed with fine elastic fibrils. Grades 4, 5, and 6 include plexiform lesions, vein-like branches of hypertrophied, usually occluded, muscular pulmonary arteries, angiomatoid lesions, and cavernous lesions (Figure 7-26). Plexiform lesions are the hallmark of grade 4 pulmonary hypertension and are formed by distention of arteries with formation of sacs with endothelial proliferation and thrombosis. Grade 6 is characterized by necrotizing arteritis with fibrinoid changes of the media with an intimal inflammatory reaction. Lesions of grades 1 and 2 and sometimes grade 3 are reversible (given surgical repair of the inciting cardiac defect). Changes found in grades 4–6 are irreversible.
FIGURE 7-26
Vascular Changes in Pulmonary Arterial Hypertension. Pulmonary arterioles/arteries from an individual with uncorrected congenital heart disease. Elastic tissue stain demonstrates adventitial fibrosis. There is also marked medial thickening and occlusion of arteriole. (grade 3 lesion). From Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill.
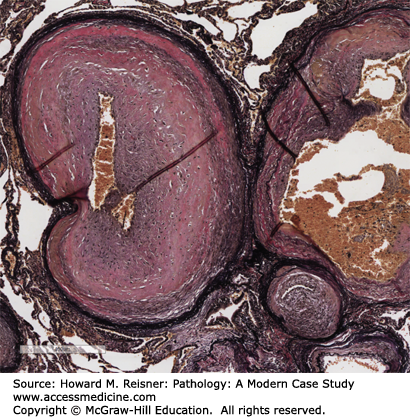
Pulmonary lung biopsies are now infrequent for grading of pulmonary arterial changes. Clinical studies, including echocardiogram and arteriogram, can provide the necessary information of pulmonary artery pressures and the possible prognosis. Additional aspects of pulmonary vascular disease are discussed in Chapter 8.
ARTERIOSCLEROSIS
Based on Greek words meaning “arteries” (arterio-) and “hardening” (sclerosis), arteriosclerosis is a pathologic process caused by thickening and loss of elasticity of the arterial walls. The three types of arteriosclerosis include
Mönckeberg medial calcific stenosis
Arteriolosclerosis (see above)
Atherosclerosis
Mönckeberg medial calcific stenosis is the calcification of the muscular media in elderly patients leading to decreased elasticity. This medial calcification does not lead to luminal narrowing or decreased blood flow. It does, however, lead to the decreased ability of muscular arteries to respond to stimuli inducing changes in resistance (Figure 7-27).
FIGURE 7-27
Mönckeberg medial calcific stenosis. Typical appearance of an artery demonstrating medial calcification. An area of calcification of the internal elastica is shown by the arrow. Reprinted from Micheletti RG1, Fishbein GA, Currier JS, Fishbein MC. Mönckeberg sclerosis revisited: a clarification of the histologic definition of Mönckeberg sclerosis. Arch Pathol Lab Med. 2008 Jan;132(1):43-7 with permission from Archives of Pathology & Laboratory Medicine. Copyright 2008. College of American Pathologists.
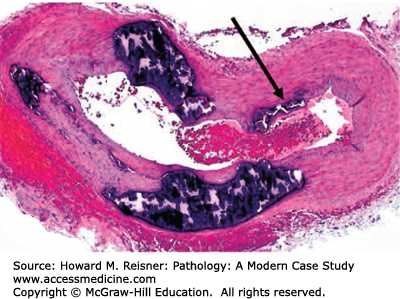
Atherosclerosis is the most common type of arteriosclerosis primarily involving end-organ arterial systems including large elastic (aorta, carotids, iliacs) and medium-sized muscular arteries (coronary arteries, femoral, popliteals). The term reflects the appearance of the atherosclerotic plaque due to accumulation of lipid, smooth muscle cells, inflammation, and connective tissue within the intima. These intimal lesions, called atheromas or atherosclerotic plaques, lead to significant morbidity and mortality, therefore, making it important to understand its etiology and pathogenesis.
The response-to-injury theory, also now known as atherothrombosis, was proposed by Ross and Fuster in the 1990s. This theory with some modification is the current, best accepted hypothesis explaining the pathophysiology of atherosclerosis. It is postulated that atherosclerosis begins with injury of the endothelium, subsequently leading to thrombosis and lipid deposition. In this theory, the interaction between the vascular wall and various physical forces regulate formation of the atherosclerotic plaque by a fibroproliferative and inflammatory response. The typical atherosclerotic plaque (atheroma) is a progressive accumulation of inflammatory cells, smooth muscle cells, lipid, and connective tissue within the intima.
The endothelium is an active and dynamic tissue that produces multiple cytokines, proteins, and tissue factors that allow normal function of the vascular system. ECs prevent thrombosis by production of a layer of heparin sulfate along the luminal surfaces. Prostacyclin also prevents thrombosis by preventing platelet aggregation (see Chapter 2). Production of endothelium-derived relaxing factor (EDRF, nitric oxide), a potent vasodilator, and potent vasoconstrictors (endothelin, serotonin, platelet-derived growth factor) allows the endothelium to maintain appropriate vascular tone. Lysis of fibrin is crucial for maintenance of blood flow and is promoted by endothelial production of plasminogen. Selective permeability of various blood components, including lipids, is also an important role of ECs.
Acute or chronic injury of the endothelium causes endothelial dysfunction. Secretion of cytokines and growth, migration, and adhesion factors occurs in the initiation and formation phase of the atherosclerotic plaque due to endothelial injury. Multiple growth factors (platelet-derived growth factor, insulin-like growth factor, α- and β- transforming growth factors, fibroblastic growth factor, and angiotensin II) are mitogens produced by activated platelets, macrophages, and dysfunctional ECs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


