INTRODUCTION
Disorders stemming from the endocrine system are among the most frequently encountered patient problems. Currently, diabetes (discussed under Pancreas) and thyroid disorders rank within the top ten most encountered diagnoses. On a broader scale, endocrinopathies cross all ages and genders and often manifest as other clinical entities that make diagnosis challenging. Because multiple organ systems are usually involved, endocrinology can seem daunting in an organ-based approach. However, it can be divided into more manageable sections by approaching the topic first by the gland involved, that is, pituitary, adrenal, or thyroid, and then from the clinical perspective of hyperfunction or hypofunction, based on the concentrations of the respective hormones or peptides secreted into the circulation. This chapter will thus be divided by system or gland and subdivided by functional defect. Diagnosis of endocrine disorders makes heavy use of clinical laboratory testing and that emphasis is reflected in this chapter.
QUICK REVIEW Basic Principles of Clinical Endocrinology Glands, Hormones, and Receptors
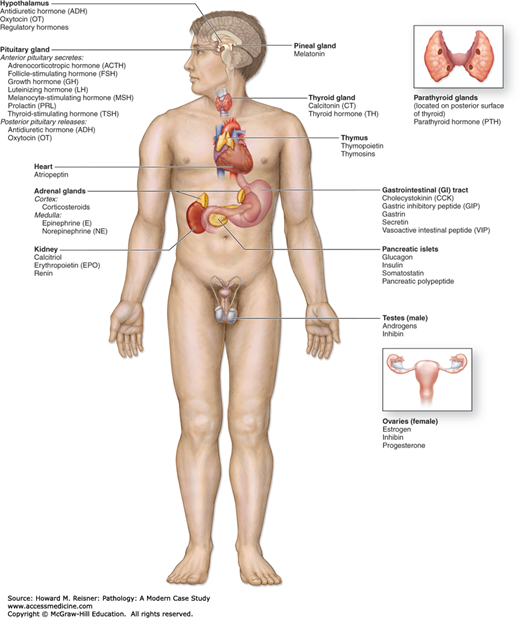
The key glands of the endocrine system include the hypothalamus, pituitary, thyroid, parathyroids, adrenals, pancreas, and gonads. These organs synthesize and secrete specific biochemical messengers, known as hormones, into the blood in a synchronized collaboration with the central nervous system (CNS) and the immune system to regulate metabolism, growth, development, and reproduction (Figure 15-1). Other tissues, such as adipose and gut, are also metabolically active and involved in these activities as well. The term hormone, derived from the Greek hormon meaning to set in motion, was chosen because these compounds act on cells some distance from their site of origin (hence the derivation of the word endocrine, from the Greek krino, to separate). Now we know that many hormones also act locally on neighboring cells at the paracrine level, and even on the very cell from which they originate in an autocrine manner.
Several classification schemes of hormones are recognized, but the simplest consists of three broad groups: peptides, steroids, and amino acid derivatives. The peptide hormones represent the largest and most diverse hormone class and include examples such as growth hormone (GH), adrenocorticotropic hormone (ACTH), and insulin. These are typically synthesized and stored for quick release into the circulation when needed, either as specific gene products or through post-translational modification of precursors and are usually water-soluble. All steroid hormones are derived from cholesterol, and as such, are lipophilic. Compounds in this class are not stored, so the rate of synthesis regulates secretion. Examples of steroid hormones include cortisol, aldosterone, and the androgens. The last group, amines, is derived from the amino acid tyrosine and includes the thyroid hormones (thyroxine (T4) and triiodothyronine (T3)) and the catecholamines (epinephrine, norepinephrine, and dopamine). Each of these compounds is stored as granules in the cytoplasm until needed.
Hormone Transport—The Role of ProteinsUpon release by the respective endocrine gland, a hormone is transported through the circulation to various tissues. En route to the target tissue the hormone is exposed to degradation by numerous proteolytic enzymes. Hormones circulate either as free (i.e., not protein bound) forms, or bound to one or more proteins. Free hormones are readily available for receptor interaction and thus exhibit the greatest biological activity, but they are most susceptible to degradation. Protein binding serves as a means of transporting hormones safely through the circulation to the target tissue and as a way of assuring sufficient hormone is available should the amount of bioavailable hormone decline. Examples of hormones and their related binding proteins are seen in Table 15-1. How tightly the hormone is bound to a protein is important since this affects the hormone’s potential to interact at the receptor, that is, its biological activity. Those that are found in the free state, along with those hormones that are weakly protein bound, are considered to have the greatest bioavailability; while hormones bound to proteins with high affinity have the least.
Peptide hormones typically circulate in an unprotected free form. They must reach their destination and act within minutes before being degraded. At the target cell surface, these compounds bind to specific receptors and in doing so activate intermediate messengers that cause a downstream response. Steroid hormones circulate in combination with a protein. As a result, they have longer half-lives (on the order of hours). Their lipophilic nature facilitates entry into the target cell in order to bind to specific intracellular receptors. The steroid–receptor complex interacts with the nucleus to elicit an alteration in gene expression and protein synthesis. Although steroids typically travel some distance from the site of origin, the importance of their actions at the local level is now recognized. Additionally, the local metabolism of hormone precursors also contributes to many of the biological effects observed. Amines circulate both free and protein bound and act via cell surface receptors (catecholamines) and through nuclear receptors (thyroid hormones).
Regulation and RhythmsA recurring theme in the following sections is the pattern of secretion observed for many hormones. Two observations must be considered when using hormone measurements in patient care. First, many display a distinctive circadian or diurnal pattern characterized by a gradual rise and subsequent fall in concentrations over the 24-hour period. The circadian pattern observed is tied to the individual’s biological clock, which in turn is synchronized to a combination of events including retinal response to light-dark, hormonal signals from sleep, awakening, and feeding, and is subject to neuronal and humoral signals originating from the suprachiasmatic nucleus as well as peripheral tissues (heart, gut, muscle). Second, although one may observe rapid rises, or pulses, in concentration to allow the body to respond quickly to changing needs, the concentrations quoted in reference ranges result from more long-term effects on the pattern of hormone secretion.
One of the most important features of the endocrine system is its ability to self-regulate by either positive or negative feedback control. Positive feedback control is stimulatory resulting in rapid changes. An example is the release of oxytocin to stimulate uterine contractions, which, in turn, stimulates the release of more oxytocin. In contrast, the overall effect in negative feedback control is inhibition. Negative feedback mechanisms exist throughout the endocrine system, and the hypothalamic–pituitary–thyroid axis is often used as an illustration. In response to the circulating levels of the key secretory hormones (thyroxin, T4 and triiodothyronine, T3) released by the thyroid, the hypothalamus and pituitary alter production of their own hormonal products, in this example, thyrotropin releasing factor (TRH) and thyroid stimulating hormone (TSH), respectively. In the case of decreased thyroid function, less T4 and T3 are released into the circulation and the hypothalamus and pituitary respond by increasing their appropriate hormones in an attempt to stimulate the thyroid. Conversely, should the thyroid become over active as in hyperthyroid states, the hypothalamus and pituitary sense the elevated T4 and T3 concentrations, and in turn, decrease production of their hormones, TRH and TSH. These events are collectively known as negative feedback and help to maintain the secretory hormone within a narrow range.
Hormonal control is further defined by the individual’s set point that some consider one’s personal reference range. The set point typically falls within the expected population reference range but is unlikely to be at the mean or median of that range. The concept is important to remember when a change in a patient’s hormone concentration is observed along with clinical signs and symptoms even if the concentration remains within the reference range.
| Protein | Hormone |
|---|---|
| Albumin | All |
| Thyroid-binding globulin (TBG) | T3, T4 |
| Thyroid-binding prealbumin (TBPA, transthyretin) | T4 |
| Cortisol-binding globulin (CBG) | Cortisol |
| Sex hormone-binding globulin (SHBG) | Estradiol, testosterone |
| Vitamin D-binding globulin (VDBG) | Vitamin D |
FIGURE 15-1
Endocrine system. The endocrine glands and major hormones they secrete are listed with their locations. In parentheses are shown other organs, including the heart, kidney, thymus, gut, and gonads, which contain endocrine cells and have important endocrine functions. In addition, many widely distributed tissues and cells throughout the body have endocrine functions but are not shown here. These include adipose cells, which secrete the hormone leptin, and vascular endothelial cells, which produce polypeptides called endothelins, which promote vasoconstriction. (Reproduced with permission from McKinley M, O’Loughlin V.D. Human Anatomy. 2nd ed. New York: McGraw-Hill, 2004 (+++Figure 20-1).)
HYPOTHALAMIC–PITUITARY AXIS
The hypothalamus serves as a link between the CNS and the endocrine system. The pituitary gland, also known as the hypophysis, is located beneath the hypothalamus in the bony sella turcica. Structurally, the pituitary can be divided into the anterior (adenohypophysis) and posterior (neurohypophysis) sections. The anterior pituitary is further subdivided into the pars distalis (accounting for the bulk of the region), pars tuberalis, and pars intermedia (Figure 15-2). Within the hypothalamus, hypophysiotropic neurons secrete specific factors that travel via the adenohypophyseal portal system to the anterior pituitary where they bind to receptors to either stimulate or inhibit the secretion of the five major anterior pituitary hormones. In addition, neurons located primarily in the supraoptic and paraventricular nuclei of the hypothalamus secrete vasopressin and oxytocin from axons extending into the neurohypophysis.
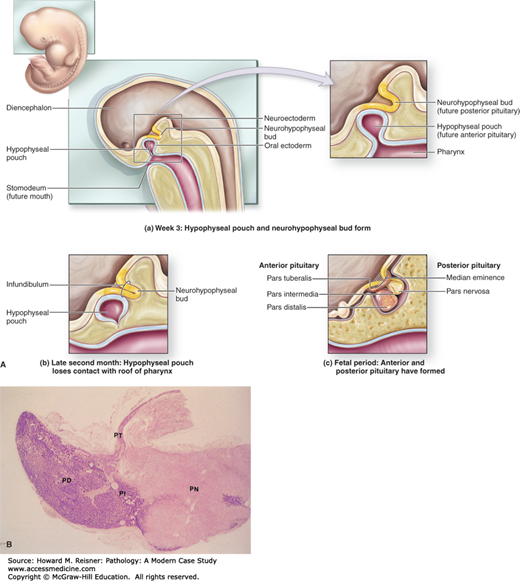
FIGURE 15-2
(A) Formation of the pituitary gland. The pituitary gland is formed from two separate embryonic structures. (a) During the third week of development, a hypophyseal pouch (or Rathke pouch, the future anterior pituitary) grows from the roof of the pharynx, while a neurohypophyseal bud (future posterior pituitary) is formed from the diencephalon. (b) By late in the second month, the hypophyseal pouch detaches from the roof of the pharynx and merges with the neurohypophyseal bud. (c) During the fetal period, the anterior and posterior parts of the pituitary complete development. (Reproduced with permission from McKinley M, O’Loughlin VD Human Anatomy. 2nd ed. New York: McGraw-Hill, 2004 (Figure 20-15).) (B) Histology of the pituitary gland. Histologically, the two parts of the pituitary gland reflect their origins, as seen in this low-magnification section of an entire gland. The infundibular stalk (IS) and pars nervosa (PN) of the neurohypophysis resemble CNS tissue, while the adenohypophysis’ pars distalis (PD), pars intermedia (PI), and pars tuberalis (PT) are typically glandular in their level of staining. X15. Hematoxylin and eosin (H&E). (Reproduced with permission from Berman B. Color Atlas of Basic Histology. 3rd ed. New York: McGraw-Hill, 2003 (Figure 17-1).)
QUICK REVIEW
Histologically, the pars distalis of the anterior lobe is composed of cords of secretory cells, that is, the somatotrophs, lactotrophs, thyrotrophs, corticotrophs, and gonadotrophs (Table 15-2). By the names alone, one can surmise that they are responsible for the synthesis and release of specific hormones: growth hormone (GH), prolactin (PRL), thyroid-stimulating hormone (TSH), adrenocorticotropin, and luteinizing hormone (LH) and follicle-stimulating hormone (FSH), respectively. Although one can use traditional histological staining methods to distinguish between the cell types, these methods cannot be used to identify the specific hormonal products. In general, however, the somatotrophs and lactotrophs exhibit acidophilic staining, while the corticotrophs, thyrotrophs, and gonadotrophs exhibit basophilic staining (Figure 15-3). The posterior lobe is nervous tissue composed of axons whose neuronal cell bodies are in the hypothalamus and modified glial cells (also known as pituicytes).
| Pituitary Hormone | Cells Responsible for Synthesis | Hypothalamus | Target Gland or Tissue | Function | |
|---|---|---|---|---|---|
| Releasing Hormones | Inhibiting Factors | ||||
| Adrenocorticotropin hormone (ACTH) | Corticotrophs | Corticotropin-releasing hormone (CRH) Arginine vasopressin (AVP) | Adrenal | Stimulation of corticosteroids and adrenal androgens | |
| Growth hormone (GH) | Somatotrophs | GH-releasing hormone (GHRH) | Somatostatin (somatotropin-release inhibiting hormone, SRIF)c | Peripheral tissue, liver | Direct and indirect (insulin-like growth factor 1, IGF-1) stimulation of growth, metabolism, homeostasis |
| Prolactin (PRL) | Lactotrophs | Oxytocin, thyrotropin-releasing hormone (TRH) | Dopamined | Mammary gland | Stimulation of lactation |
| Thyrotropin or thyroid-stimulating hormone (TSH)a | Thyrotrophs | TRH | Somatostatin | Thyroid | Stimulation of thyroid hormone release |
| Luteinizing hormone (LH)a | Gonadotrophs | Gonadotropin-releasing hormone (GnRH)b | Ovary, testis | Stimulation of estrogen and testosterone production | |
| Follicle-stimulating hormone (FSH)a | Gonadotrophs | GnRHb | Ovary, testis | Regulation of theca and Sertoli cell function | |
| Human chorionic gonadotropin (hCG)a | Gonadotrophs | GnRH | unknown | unknown | Differs from placental and trophoblastic isoforms. The role of pituitary hCG is unknown—it may serve to facilitate some of the LH functions |
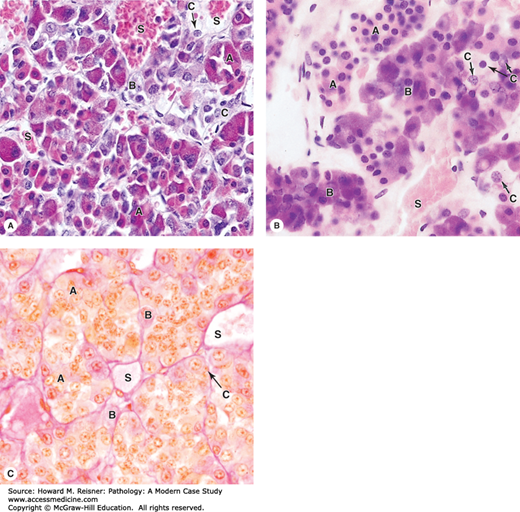
FIGURE 15-3
Pars distalis: Acidophils, basophils, and chromophobes. (A,B). Most general staining methods simply allow the parenchymal cells of the pars distalis to be subdivided into acidophil cells (A), basophils (B), and chromophobes (C) in which the cytoplasm is poorly stained. ×400. H&E. (B) Gomori trichrome provides similar information. ×400. Cords of acidophils and basophils vary in distribution and number in different regions of the pars distalis, but are always closely associated with capillaries and sinusoids (S) in the second capillary plexus of the portal system. The vascular plexus carries off secreted hormones into the general circulation. Specific acidophil or basophil cells can be identified immunohistologically with antibodies against their hormone products. Chromophobes are less numerous and represent various undifferentiated parenchymal cells. Their number and density also vary in different regions. (Reproduced with permission from Mescher AL. Junqueira’s Basic Histology: Text and Atlas. 12th ed. New York: McGraw Hill Lange, 2010:353 (Figure 20-6).)
TROPIC HORMONES OF THE ANTERIOR PITUITARY
The hormones of the anterior pituitary include GH, PRL, ACTH, TSH, LH, and FSH. Abnormalities associated with GH and PRL are of particular significance in terms of anterior pituitary pathology.
GH is the most abundant hormone produced by the anterior pituitary. The hormone is a relatively small, single-chain peptide that circulates in both free and protein-bound forms in about a 50-50 ratio. Hypothalamic growth hormone releasing hormone (GHRH) stimulates release, while somatotropin-release inhibiting factor (SRIF, somatostatin) inhibits release. The actions of GHRH, that is, release, are now thought to dominate since disruption of the pathway leads to GH deficiency. Negative feedback through a short feedback loop also plays a role as GH itself stimulates the hypothalamus to produce SRIF.
GH deficiency is a problem encountered in children who have not achieved their normal stature. This excludes children who are genetically predisposed to being short statured, who are malnourished, or who have an underlying systemic illness.
The terms hypopituitarism and GH deficiency are often used interchangeably because pituitary dysfunction rarely occurs without GH deficiency. Hypopituitarism has many causes, as seen in Table 15-3, and the presentation varies depending on the age of the patient. Disorders of the hypothalamus may cause GH deficiency by impairing GHRH secretion, whereas lesions of the pituitary or the pituitary stalk may directly cause GH deficiency. Unfortunately, “idiopathic hypopituitarism” is the most frequent cause of GH deficiency in children and may occur as a result of trauma during birth or in the perinatal period. For others, as yet unrecognized genetic mutations are likely. In many cases, nutritional, psychological, or pharmacological causes are suspected.
| Pituitary Diseases | |
| Genetic etiologies | Aplasia and hypoplasia due to mutations in a pituitary transcription factors, such as Prop-1 and Pit-1 Isolated GH deficiency due to mutation in the GH-N gene |
| Intrasellar tumors | Craniopharyngiomas, adenomas, and metastatic tumors |
| Nontumorous destruction | Infarction associated with trauma, infection, head irradiation, postpartum necrosis (Sheehan syndrome) |
| Disorders of the Hypothalamus | |
| Idiopathic | Often associated with birth trauma and other forms of perinatal injury |
| Familial forms | Midline central nervous system (CNS) and facial development defects: septo-optic dysplasia, holoprosencephaly, cleft lip or palate, single upper central incisor |
| Infections and inflammatory diseases | Tuberculosis, sarcoidosis |
| Histiocytosis | |
| Hypothalamic tumors | Craniopharyngiomas, neurofibromas, gliomas, germinomas, metastatic tumors to the hypothalamus |
| Functional disorders | Psychosocial dwarfism, anorexia nervosa |
| Disorders of Growth Hormone (GH) Responsiveness (GH high, IGF-I low) | |
| Receptor abnormalities | Laron dwarfism (GH receptors mutations), pygmies |
| Biologically inactive GH | |
| Protein-calorie malnutrition | |
Most defects in multiple pituitary hormone secretion are related to compressive or other tissue destructive causes including CNS tumors (craniopharyngioma being a notable example), neurological insults, and cranial radiotherapy. In such cases the cells responsible for the hormone synthesis will usually cease, functioning in a specific order. Loss of GH typically occurs first followed by loss of LH/FSH, then loss of TSH, and finally ACTH.
Congenital deficiencies of pituitary hormones are uncommon (1 in 50,000 births). They may affect pituitary gland development and be associated with hyposecretion of multiple pituitary hormones (combined pituitary hormone deficiency CPHD or panhypopituitarism). Congenital CPHD results from mutations in genes for developmental factors necessary for pituitary gland maturation. Most of these genes affect the anterior pituitary. Mutations in two transcription factors (POUF-1/PIT-1 and PROP1) are associated with CHPD (the latter accounting for about 30% of cases) in which GH as well as PRL and TSH are deficient.
Isolated growth hormone deficiency (IGHD) is associated with mutations in the growth hormone gene (GH1) and in the GHRH receptor gene. The most severe form (Type 1A) is associated with deletion of the GH1 gene. Most cases of IGHD are idiopathic (not currently associated with a known genetic defect). Genetic defects in hypothalamic factors have also been associated with congenital pituitary defects. Therapy relies on recombinant human GH. Patients with Type 1A disease may become refractory to therapy as a result of developing neutralizing antibodies to the recombinant protein.
WHAT WE DO Diagnosis of Hypopituitarism and GH Deficiency in Children
In the newborn or neonatal period, IGHD and even panhypopituitarism may be difficult to identify. Growth is usually not affected during this period and there may be few, if any, symptoms with IGHD. A significant clue of panhypopituitarism during this period, however, is the occurrence of episodes of hypoglycemia due to the combined deficiency of cortisol and GH. In childhood, a decrease in linear growth velocity is a hallmark feature of both IGHD and panhypopituitarism. This is often apparent after 6 months of life, but can occur at any age. These children are often modestly overweight and look younger than their age due to immature facial bone structure. Without treatment, those with panhypopituitarism will not progress through puberty and will eventually exhibit signs and symptoms of TSH and/or ACTH deficiency as the disease progresses.
A single GH measurement is not an optimal measure to assess deficiency. In children, it is better to assess the response of the pituitary to a stimulus such as the tests shown in Table 15-4 (GH stimulation tests). IGF-1 contributes to GH regulation by directly stimulating the hypothalamus to release SRIF. Because IGF-1 has a much longer half-life compared with GH, it is useful as a substitute for GH testing when evaluating patients. Although GH-deficient patients will usually have corresponding low IGF-1 concentrations, a low IGF-1 is not specific for GH deficiency but may be related to poor nutrition, renal disease, hypothyroidism, or psychological disorders.
Diagnosis of Hypopituitarism in AdultsThe presentation of hypopituitarism in an adult depends on the hormone(s) that are deficient, the magnitude of the deficiency, and the rapidity of occurrence. For example, symptoms, including those of ACTH deficiency, may occur quite rapidly following infarction or postpartum necrosis (Sheehan syndrome).
Progression of hypopituitarism may be insidious following cranial irradiation and symptoms may be subtle for many years. Adults typically are diagnosed after they encounter symptoms related to secondary hypogonadism (amenorrhea for women and impotence for men). With progression or involvement of the thyrotrophs, signs and symptoms of secondary hypothyroidism may develop. In some cases, the patient is diagnosed when symptoms of secondary hypoadrenalism develop and they are unable to handle a stress—in severe cases, a life-threatening adrenal crisis may ensue causing vascular collapse.
Testing will show low tropic hormones from the pituitary and low secretory hormones from the endocrine gland, for example, a low TSH with a low thyroxine. Additional provocative testing may be needed to sort through apparent mild or subtle deficiencies. These include the use of GnRH to test FSH and LH reserve, ACTH to test adrenal reserve, TRH to distinguish pituitary from hypothalamic causes of abnormal thyroid function, and so on.
CASE 15-1
A 21-year-old female is found unresponsive at home. In the emergency room, she is found to have altered metal status, is afebrile with a pulse rate of 50 beats per minute, respiration 12 per minute, and blood pressure is 100/60 mmHg. Stat laboratory results (with reference ranges) include sodium 134 mmol/L (135–145), potassium 4.0 mmol/L (3.5–5.0), chloride 110 mmol/L (98–107), and a whole blood glucose (point of care) 40 mg/dL (65–179). (Quick review of these laboratory values: the most significant result is the critically low glucose, consistent with hypoglycemia. The sodium is mildly decreased and the chloride is mildly increased. As part of the exercise, you may want to stop and think about the causes of these changes.)
She became responsive with emergent treatment that included intravenous administration of 50% dextrose. Her family reports that she gave birth to her third child 4 months earlier. Since that time she has complained of fatigue, malaise, and weakness, which was attributed to postpartum depression and the care of three children plus full-time employment. They also note, however, that she has had episodes of vomiting, abdominal pain, and diarrhea. She does not use alcohol or drugs, is not diabetic, and until the delivery was in good health and active.
The patient’s hypoglycemia may be a life-threatening event. Many times its occurrence is related to the effect of a medication or alcohol use, but other causes include liver disease, uremia, neoplasms (insulinoma, for example), metabolic disorders such as glycogen storage diseases, and endocrinopathies such as hypoadrenalism, hypopituitarism, and glucagon deficiency. Additional testing reveals a TSH of <0.1 μIU/mL (0.4–4.5), cortisol less than 1.5 μg/dL (5–20), FSH 1.0 μIU/mL (1.5–12), PRL 1.3 ng/mL (3–20).
With these results, panhypopituitarism resulting from postpartum infarction of the anterior pituitary, also known as Sheehan syndrome, is suspected. This condition was first described in 1937 by a British pathologist. Improved obstetrical care has reduced the frequency in developed countries, but it still occurs. The blood flow to and within the anterior pituitary is among the highest of any other organ. During pregnancy, the pituitary enlarges and becomes vulnerable to ischemia. In the event of severe postpartum hemorrhage, the resulting systemic hypotension may lead to vasospasm and pituitary infarction.
Excessive GH production leads to acromegaly in adults and gigantism when it occurs in children. At least 90% of all cases of acromegaly are associated with pituitary adenomas. Neoplasms of the pituitary are almost invariably benign and common. Autopsy and radiographic studies suggest that between 15% and 20% of normal persons have unapparent tumors; symptomatic tumors are much less common (0.1% in community-based studies). Although benign, pituitary adenomas may have serious clinical consequences involving mass effects (such as headache, cranial nerve palsies, bitemporal hemianopia, and visual defect in the temporal half of each eye’s visual field), panhypopituitarism and ultimately destruction of the sella as a result of pressure-related damage. Symptoms relating to mass effects are usually associated with macroadenomas (larger than 1cm in diameter). Rapid expansion of a macroadenoma as a result of infarction and/or hemorrhage may result in pituitary apoplexy, the acute onset of symptoms related to mass effects requiring prompt surgical intervention. Hyperfunction of the pituitary, and in particular excess of a single hormone, is (as noted above) most often the result of a functional (hormone secreting) adenoma, which comprises about 80% of pituitary adenomas.
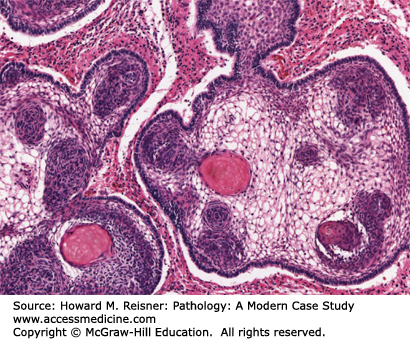
Most adenomas are derived from the anterior pituitary and are monoclonal, demonstrating sheets or cords of uniformly stained cells (Figure 15-4). Twenty-five percent of pituitary adenomas are lactotrophs (prolactinomas) and are the most common type. Somatotropic (GH secreting) adenomas are relatively uncommon (5–15% of adenomas). Twenty-five percent of GH secreting adenomas also secrete PRL. A proportion of such multiple hormone secreting tumors is polyclonal. Monoclonal, pluripotent primitive stem cell-derived tumors occur, are aggressive, and are associated with gigantism in teenagers. Pituitary adenomas may be sporadic or familial and details of the molecular pathology of such are available.
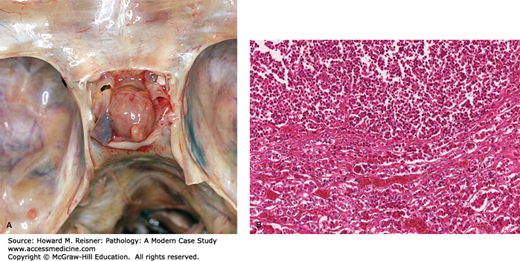
FIGURE 15-4
Pituitary adenoma. (A) Gross photograph of organ exposed in sella turcica of sphenoid. (B) Photomicrograph of pituitary pars dorsalis demonstrating adenoma in upper half of image. Note uniform population of acidophilic cells in contrast to mixed population in the normal area (bottom half of the image). ((4a) Contributed with permission of Clay Nichols, MD NC Medical Examiners Office.)
The patient who has acromegaly has a distinctive physical appearance due to acral growth of flat bone that leads to large, broad features of the hands, feet, and nose. The skin becomes thickened due to excess collagen. Physical examination will reveal a generalized enlargement of the organs. These disorders can progress slowly and not be recognized for some time. These patients usually have elevated GH concentrations that do not suppress (see below). Because (as noted above) IGF-I concentrations are directly influenced by GH and are not subject to rapid fluctuations, they are now used in the diagnosis of these patient suspected to have excess GH production. IGF-I concentrations also correlate well with clinical severity of the disease. Treatment is complex and includes radiosurgical approaches or the use of pharmacological agents including somatostatin receptor ligands (somatostatin analogues) and GH receptor antagonists.
PRL is a 198 amino acid protein produced by the lactotrophs. As the name of these cells suggest, PRL controls initiation and maintenance of lactation providing the breast tissue has been primed by estrogens, progestins, corticosteroids, thyroid hormone, and insulin. Regulation of PRL is unique in that primary control is inhibitory rather than stimulatory. Dopamine appears to be the main PRL inhibitory factor, but there may be others. Several PRL releasing factors have been identified including thyrotropin-releasing hormone (TRH), vasoactive intestinal peptide, and estrogen. PRL deficiency is important during the postpartum period because it is necessary for lactation. In such cases the PRL deficiency may be related to Sheehan syndrome (see Case 15-1). Outside of this period and in males, the impact is not well defined.
The diagnosis of hyperprolactinemia, the most common hypothalamic–pituitary disorder, can be quite challenging as there are multiple etiologies for the disorder. Mild elevations in PRL may be seen in chronic renal failure, following breast stimulation, in primary hypothyroidism, in empty sella syndrome, and in response to many drugs (antihypertensives, phenothiazines, morphine, methyldopa, and some antidepressants). Even mild hyperprolactinemia can inhibit ovulation and lead to infertility. Patients who are found to have a macroprolactinoma can have very elevated PRL concentrations. Tumors of the lactotrophs vary considerably in size and in their production of PRL. However, many prolactinomas are less than 1 cm in diameter and grow slowly thus permitting careful monitoring if detected. The elevated PRL inhibits the secretion of gonadotropin releasing hormone (GnRH) that leads to ovarian dysfunction, amenorrhea, and galactorrhea. This syndrome is the most common presentation of patients with functioning pituitary adenomas. Nonsurgical treatment involves the use of dopamine D2 receptor agonists such as bromocriptine and cabergoline, which directly inhibit hormone secretion.
WHAT WE DO
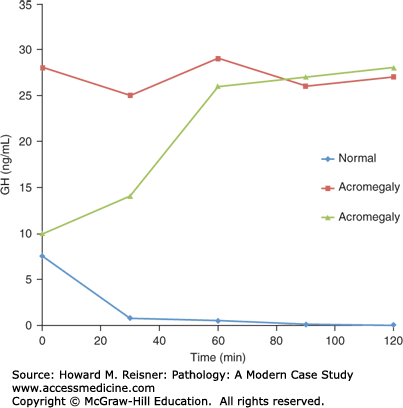
The diagnosis of acromegaly is confirmed by performing an oral glucose tolerance test and noting the failure of the GH concentration to suppress as seen in Figure 15-5. A normal response (diamonds) is a suppressed serum GH to <1 ng/mL. In contrast, all acromegaly patients respond abnormally showing either (1) a lack of suppression, (squares) or (2) a paradoxical increase (triangles), a response seen in ~20% of cases for unknown reasons.
CASE 15-2
ED is a 42-year-old female who begins to experience frequent headaches that she thinks are related to a need for new glasses. On a routine examination by her optometrist, she is found to have a visual field defect and was immediately referred for workup. He immediately refers ED to you. During her history and physical, she reports she has had irregular menses for a number of years but few other complaints or problems. MRI reveals a tumor of approximately 1.2 cm in size within the sella. Laboratory testing is within normal limits with the exception of PRL, which is elevated at 1152 ng/mL. In the absence of PRL-stimulating drugs or a recent pregnancy, the increased PRL concentration suggests the tumor is a prolactinoma.
Besides PRL and other hormones discussed earlier, additional tropic hormones secreted by the anterior pituitary include ACTH, TSH, LH, and FSH and are discussed as follows.
ACTH is a product of the corticotrophs. This 39 amino acid polypeptide fragment originates from a larger precursor molecule, proopiomelanocortin (POMC) that also gives rise to β-lipotropin. Corticotropin-releasing hormone (CRH) and proinflammatory cytokines stimulate release of the peptide in response to biorhythms and various stimuli. ACTH synthesis can also be stimulated by high concentrations of antidiuretic hormone (ADH) interacting with the vasopressin type 3 receptors on the corticotrophs. ACTH exerts a number of effects on the adrenal cortex including the incorporation of lipoprotein-bound cholesterol into the gland, stimulation of RNA and protein synthesis, and promotion of cell differentiation. Most importantly, it is the primary regulator of the secretion of cortisol and androgenic steroids by the adrenal cortex.
Because of this close relationship, ACTH and cortisol follow very similar patterns of release. During a 24-hour period, maximal (peak) concentrations are achieved between 0400 and 0800 with the lowest (trough) concentrations seen approximately 12 hours later in response to sleep-wake cycles. Individuals who have altered hours of sleep and wakefulness such as those who work the midnight shift, for example, have a shift, even a reversal, in their diurnal rhythm. In addition to the characteristic diurnal pattern, episodic increases in concentrations are associated with various stressors.
LH and FSH are synthesized and released from gonadotrophs located in the anterior pituitary in response to GnRH. GnRH is secreted by the hypothalamus in a pulsatile manner in response to estrogen in the female and testosterone in the male. Biochemically, LH and FSH are glycoproteins consisting of an α- and β-subunit. The α-subunit that is involved in receptor binding is identical in structure to the α-subunit of both TSH and human chorionic gonadotropin (hCG). The unique β-subunit of each provides immunospecificity and hormonal activity. Both subunits are necessary for full biological activity. Once released from the gonadotrophs, LH and FSH travel through the circulation to the gonads. In the female, FSH stimulates the growth and maturation of ovarian follicles, stimulates estrogen secretion, and promotes endometrial changes of the proliferative phase of the menstrual cycle. LH acts with FSH to promote ovulation and secretion of androgens and progesterone. LH also initiates and maintains the secretory phase of the menstrual cycle and contributes to the formation of the corpus luteum. For each hormone, serum concentrations in women vary with the menstrual cycle and increase postmenopause. In the male, FSH stimulates spermatogenesis through its actions on the Sertoli cells, while LH stimulates development and function of Leydig cells in the testes.
Because of the actions of LH and FSH on gonadal function, gonadal failure is often the earliest symptom of pituitary insufficiency. The most direct approach to ruling out primary gonadal insufficiency from pituitary insufficiency is to measure the pituitary gonadotropins. In these cases, depressed LH and FSH concentrations are suggestive of pituitary insufficiency, while elevated concentrations rule out pituitary deficiency.
TSH (thyrotropin) stimulates growth and vascularity of the thyroid gland, growth of the follicular cells, and promotes most of the steps in thyroid hormone synthesis. TSH release is regulated by hypothalamic TRH and the thyroid hormones, T3 and T4. TSH is a 31-kDa glycoprotein composed of two subunits: the α-subunit shared with other glycoproteins (LH, FSH, and hCG) and a unique β-subunit that confers specificity. TSH will be discussed in greater detail in the thyroid section of the chapter.
hCG is not typically thought of as a pituitary hormone, but in fact, small amounts of this hormone can be produced by the gonadotrophs, most often during the peri- and postmenopausal periods. The pituitary-derived hCG concentrations found in the serum and urine are low (usually <15 IU/L), but may raise concerns of pregnancy or gestational trophoblastic disease. An elevated FSH is helpful in the immediate assessment of a patient to rule out pregnancy prior to an urgent procedure. hCG of pituitary origin should suppress with 2 weeks of estrogen replacement. If the low hCG levels persist, trophoblastic disease must be considered.
TUMORS AFFECTING PITUITARY FUNCTION
Adenomas are by far the most common tumors of the pituitary. These have already been discussed in the section “GH Excess and Pituitary Adenomas.”
Craniopharyngiomas are among the more common causes of panhypopituitarism in children. Adamantinomatous (demonstrating areas of calcification) craniopharyngiomas are most common in youth. Such tumors are commonly associated with mutations in the β-catenin gene, an activator of the Wnt signaling pathway. Squamous papillary tumors are more common in adults and are not associated with a known genetic lesion (Figure 15-6). In either case, craniopharyngiomas are composed of epithelial cells that proliferate and occupy space in and above the sella turcica, encroaching on adjacent pituitary and hypothalamic tissue. Although the tumors are defined as benign by histology, they grow slowly in an invasive pattern and tend to recur, particularly when complete surgical removal is problematic. The tumors proliferate such that symptoms manifest in the first few years of life, though there are cases in which the lesion is not apparent clinically until late in life.
IN TRANSLATION
Forty percent of sporadic GH secreting tumors have an activating mutation in the G protein subunit α-gene (GNAS or gsp) that results in constitutively elevated levels of cAMP. Somatic mosaicism for such activating GNAS mutations result in McCune–Albright syndrome (MAS), which is characterized by bone abnormalities (polyostotic fibrous dysplasia), dermal café-au-lait spots, and a spectrum of endocrine disorders notably including acromegaly. Although MAS is rare, somatic activating mutations for GNAS are common in sporadic pituitary adenomas (as noted above). Hence, such mutations are likely to play a role in adenoma generation. A number of familial inherited disorders are also associated with pituitary adenomas. The most important of which is multiple endocrine neoplasia syndrome type 1 (MEN1). MEN1 is an autosomal dominant disorder associated with mutation in the MEN1 gene, which encodes the nuclear protein menin. MEN1 functions as a tumor suppressor and tumors are associated with loss of heterozygosity (LOH) at the locus. MEN1 is characterized by the formation of adenomas in multiple endocrine organs (including the parathyroid gland in 90% of patients and the pituitary gland in 10–60% of patients). In addition, endocrine tumors of the thyroid gland, adrenal cortex, and pancreas are common. Nonendocrine tumors include lipomas, meningiomas, facial angiofibromas, and carcinoids at various sites. MEN1 is responsible for 3% of pituitary adenomas, which when functional, most often secrete GH and PRL. Progression to malignancy, although uncommon, has been reported to occur.
Pituitary carcinomas are exceedingly rare constituting no more than 0.2% of all pituitary tumors. They are defined by their ability to metastasize beyond the pituitary to the cerebrospinal space or extracranial regions. Most pituitary carcinomas are functional with nearly half the tumors secreting ACTH, which is rarely secreted by adenomas.
POSTERIOR PITUITARY
The posterior pituitary is predominately neural tissue derived from downward evagination of the neural tube. Most, but not all cells, are located in the supraoptic and paraventricular nuclei and are responsible for the synthesis of oxytocin and ADH (also referred to as vasopressin, arginine vasopressin, AVP). As with other hormones, oxytocin and ADH originate from two precursor proteins, that is, prooxyphysin and propressophysin.
Oxytocin is released in response to vaginal and lower uterine distention during labor. Upon release, oxytocin stimulates contractions of the uterine smooth muscle. In addition, oxytocin is released in response to suckling in order to stimulate the myoepithelial cells of the mammary alveoli thus causing milk ejection and efficient emptying of the breasts.
The release of ADH is mediated by osmoreceptors in the hypothalamus that respond to plasma osmolality. A decrease in plasma volume, stress, nicotine, morphine, and barbiturates can also stimulate release. The primary role of ADH is to reduce water excretion by concentrating urine through its actions on the distal tubules and medullary collecting ducts. Disorders of ADH can be divided into hypofunction (polyuric states and diabetes insipidus, DI) and hyperfunction (syndrome of inappropriate ADH secretion, SIADH).
While “normal” urine output is not definable, any patient who excretes more than 2.5–3.5 L/day warrants investigation for polyuria. When there is insufficient ADH, the renal cells become impermeable to water and reabsorb little of the dilute filtrate entering from the proximal nephron. As a result, large volumes of very dilute urine are excreted. The range of impairment is extremely variable—in some, the effect is so intense that the person excretes 5–10 L/day. Usually, the urine-specific gravity is close to that of water (approximately 1.000) and the osmolality is also decreased to less than 300 mOsm/kg. Hypothalamic diabetes insipidus (HDI) occurs when there is insufficient ADH produced (also called neurohypophyseal or central DI). This is most commonly caused by trauma to the pituitary resulting from tumors or surgery. DI may also be related to renal disease or behavioral disturbances. The latter is associated with excessive fluid consumption (psychogenic polydipsia).
WHAT WE DO
Laboratory studies for polyuria include a random serum and urine osmolality, urine glucose, fasting serum glucose, and serum sodium. A 24-hour urine collection is necessary to measure and document urine volume. Most healthy individuals will excrete less than 2.5 L/day. If serum osmolality ≥295 mOsm/kg, serum sodium ≥145 mmol/L, or glucose is >100 mg/dL the polyuria is most likely due to a condition other than diabetes insipidus-diabetes mellitus, for example. The combination of a urine osmolality less than 300 mOsm/kg with a serum osmolality above 300 mOsm/kg or with hypernatremia suggests DI is likely. If both serum and urine osmolality are decreased, psychogenic polydipsia should be considered.
The syndrome represents the autonomous, sustained production of ADH in the absence of known stimuli. SIADH is associated with a broad range of potential etiologies including ectopic hormone production by neoplasias (including carcinomas and carcinoids), drug effects, pulmonary disease, and a host of neurological/psychiatric disorders. The diagnosis is mainly one of exclusion after investigating other causes that stimulate ADH production, for example, cardiac, hepatic, renal, thyroid, and adrenal failure, or medications. ADH levels increase disproportionately to (low) serum osmolality and to a normal or increased plasma volume. Water retention results in hyponatremia and serum hypo-osmolality without edema.
THE ADRENAL GLANDS AND THEIR HORMONES
The two adrenal glands are situated retroperitoneally at the upper pole of each kidney. Each gland consists of a reddish-brown medulla surrounded by a yellow to yellow-brown cortex. The lipid and steroid contents of the cortex can be judged macroscopically by this yellow color, with brown indicating lipid and therefore steroid depletion. In the healthy adult, each gland weighs approximately 4 g. With associated pathological conditions, adrenal size may vary considerably. For example, gland weights are usually somewhat less in conditions of deficiency, while weights may be increased as much as fourfold in conditions of excess (Figure 15-7).
FIGURE 15-7
Adrenal gland. Inside the capsule of each adrenal gland is an adrenal cortex, formed from embryonic mesodermal cells, which completely surrounds an innermost adrenal medulla derived embryologically from neural crest cells. Both regions are very well vascularized with fenestrated sinusoidal capillaries. Cortical cells are arranged as three layers: the zona glomerulosa near the capsule, the zona fasciculata (the thickest layer), and the zona reticularis. (Reproduced with permission from McKinley M, O’Loughlin VD. Human Anatomy. 2nd ed. New York: McGraw-Hill, 2004 (Figure 20-13C,D).)
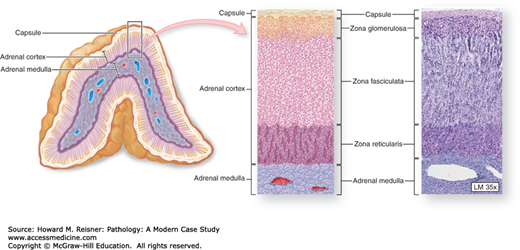
Each adrenal gland is 90% cortex that is histologically divided into three distinct zones: the outer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


