INTRODUCTION
WHAT WE DO
Hematopathology is one of the most diverse areas of pathology, since it represents a hybrid discipline involving clinical pathology (i.e., laboratory medicine) and anatomic pathology (surgical pathology of lymph nodes and extranodal tissues involved with hematolymphoid disorders). This field of pathology also uses cytomorphology and histology in combination with numerous ancillary tools (i.e., enzyme cytochemical and immunohistochemical staining, flow cytometric immunophenotyping, and cytogenetic and molecular techniques) in diagnosing and prognosticating hematolymphoid disorders. In some instances, the appropriate diagnosis of the hematolymphoid disorder also relies heavily on clinical and radiologic data.
For example, acute promyelocytic leukemia (APL) (as described later in this chapter) has characteristic, unique cytomorphological, enzyme cytochemical, and flow cytometric immunophenotypic features that are defined by the ever-present t(15;17)(q24;21) or similar variant. This translocation may be detected most rapidly by fluorescence in situ hybridization (FISH) performed in the cytogenetics laboratory. It is important to recognize this subtype of acute myeloid leukemia (AML) as soon as possible, since it is often associated with disseminated intravascular coagulation (DIC) and has a specific therapy (different from all other forms of AML).
In addition, for example, evaluation of plasma cell proliferations relies heavily on cytomorphological and histological features in combination with immunohistochemical and in situ hybridization studies for clonality. However, even with these techniques, the appropriate classification (and prognostication) of the plasma cell dyscrasia (PCD) must be based on close integration of the clinical and radiologic data (as well as cytogenetic and molecular data, respectively) in each case, as described again later in this chapter.
DISORDERS OF ERYTHROID CELLS
Group of erythroid disorders, due to nonneoplastic and neoplastic etiologies, resulting in anemia.
Anemias represent a decrease in red cell mass or hemoglobin (Hgb) concentration, and often manifest with clinical features related to overall inadequate oxygen transport and/or volume depletion. The diverse underlying pathophysiologic mechanisms can be conceptually divided into three broad categories: decreased red cell production, increased destruction or turnover, and blood loss. In principle, the latter is most straightforward, but the source of bleeding is not always easily identified. Causes of decreased production, also referred to as hypoproliferative anemias, and increased destruction, can be related to a variety of neoplastic and nonneoplastic etiologies. Common causes of anemia are listed in Table 14-1. A thorough history and physical examination along with an appropriately directed laboratory investigation are paramount to properly identifying the cause and guiding therapy.
| Microcytic | Normocytic | Macrocytic |
|---|---|---|
| Iron deficiency anemia | Blood loss | B12/folate deficiency |
| Thalassemia | Erythropoietin deficiency | Myelodysplastic syndrome |
| Sideroblastic anemias | Anemia of chronic disease | Liver disease |
| Lead poisoning | Hypothyroidism | |
| Anemia of chronic disease | Chronic alcohol ingestion |
Automated peripheral blood (PB) analyzers in clinical laboratories provide measurements of Hgb and hematocrit (HCT) concentration, or the percent of blood volume composed of red blood cells. However, these instruments output other important red cell characteristics, such as the mean corpuscular (or cellular) volume (MCV) and red cell distribution width (RDW). The MCV can be used to further define anemias as microcytic, normocytic, or macrocytic if the mean red blood cell volume is less than, equal to, or greater than normal, respectively. Ultimately, a manual review of the PB smear may provide additional information and insightful clues as to the underlying cause of anemia.
CASE STUDY: Iron-Deficiency Anemia
Iron-deficiency anemia is a microcytic anemia due to a deficiency of iron, which is essential for the synthesis of Hgb
A 35-year-old woman presents to her primary physician with a several month history of slowly progressive fatigue.
CBC Data
WBC count: 10,900/μL
Hgb: 9.3 g/dL
MCV: 62
RDW: 19%
Platelet count: 399,000/μL
The PB smear (Figure 14-1A) demonstrates a hypochromic and microcytic anemia with numerous platelets in the background. The patient has a long-standing history of irregular and heavy menstrual cycles and previously identified uterine leiomyomata. Further laboratory studies reveal the following results:
Serum iron: 5 μg/dL (26-170 μg/dL)
Total iron binding capacity: 750 μg/mL (262-474 μg/dL)
Ferritin: 4 ng/mL (12–160 ng/mL)
The clinical and laboratory findings are classic for iron-deficiency anemia, likely resultant from chronic blood loss. Iron deficiency is a common cause of anemia worldwide, resulting from insufficient iron for appropriate Hgb synthesis.
In addition to the microcytic anemia, patients with iron deficiency can present with thrombocytosis and leukocytosis (as seen in this case). The serum iron levels are typically low, although may reflect recent iron intake rather than adequate iron stores. Total iron binding capacity is increased and the iron binding protein, transferrin, is similarly elevated. Levels of the intracellular iron regulatory protein, ferritin, effectively relate to the total systemic iron stores. Although rarely biopsied if clinically straightforward, cases refractory to iron replacement may necessitate a bone marrow (BM) evaluation. The findings in the BM are not specific and vary in severity (Figure 14-1B).
FIGURE 14-1
Iron deficiency anemia. The PB smear (A, Wright stain, ×1000) demonstrates a hypochromic microcytic anemia. A circulating lymphocyte, present in the center of the image, is available for size comparison. The Wright-Giemsa-stained BM aspirate (B, ×600) shows a normocellular BM with relative erythroid hyperplasia. Erythropoiesis is typically morphologically normal, although some dysplastic features may be identified. The iron stains would reveal absence of adequate iron stores.
CASE STUDY: Refractory Anemia [Refractory Anemia with Ring Sideroblasts (RARS)]
Refractory anemia falls within the group of myelodysplastic syndromes (MDS) due to ineffective erythropoiesis.
A 79-year-old man presents with a 4-year history of a macrocytic anemia, who is now transfusion dependent.
CBC Data
WBC count: 7200/μL
Hgb: 9.3 g/dL (after transfusion)
MCV:100
RDW: 22.3
Platelet count: 275,000/μL
The BM aspirate and biopsy are hypercellular and demonstrate an erythroid hyperplasia with significant erythroid hyperplasia and dyspoiesis of the erythroid lineage (Figure 14-2A). Stains for iron demonstrate increased iron stores and numerous ring sideroblasts (Figure 14-2B). These clinical and morphologic features are consistent with myelodysplasia, specifically RARS.
MDS represent a heterogenous group of hematopoietic neoplasms with a variably increased risk of progression to AML. These malignancies typically present with otherwise unexplained PB cytopenia(s) that are a consequence of ineffective hematopoiesis. RARS represents a common form of MDS specifically affecting erythropoiesis. In contrast to other forms of MDS, refractory anemia has a relatively indolent clinical course and a low risk of progression to acute leukemia.
FIGURE 14-2
RARS. As is typical for most cases of MDS, the BM aspirate (A, Wright stain, ×600) is hypercellular. The erythroid series shows dyspoietic features including irregular nuclear contours with nuclear blebbing (arrows) and megaloblastoid changes. The granulocytic and megakaryocytic lineages are, by definition, morphologically normal. Iron stain of the aspirate (B, ×1000) shows abundant iron (blue) and many ring sideroblasts (arrows), representing mitochondrial iron deposition along the nuclear border.
Group of erythroid disorders due to nonneoplastic and neoplastic etiologies resulting in polycythemia (Table 14-2).
CASE STUDY: Copper-Deficiency Anemia HUNTING FOR ZEBRAS
Copper deficiency is rare and may cause sideroblastic anemia (case of copper-deficiency anemia).
The patient is a 12-year-old boy with a history of microcytic hypochromic anemia.
CBC Data
WBC count: 3000/μL
Hgb: 8.0 g/dL (after transfusion)
MCV: 57
Platelet count: 250,000/μL
Laboratory testing and supplementation have excluded iron deficiency.
Although B12 and folate deficiency are most commonly considered, a variety of nutritional deficiencies can clinically manifest with signs and symptoms of anemia. Copper deficiency is a rare cause of anemia, and can present with neurologic manifestations. Although hereditary and several acquired causes have been identified, the etiology remains uncertain for many patients. PB manifestations are diverse and include micro-, normo-, and macrocytic anemia, leukopenia with neutropenia, and less commonly, thrombocytopenia. The BM in these patients demonstrates erythroid dyspoiesis, in many cases morphologically indistinguishable from primary MDS. Iron stains highlight ring sideroblasts, thus making copper deficiency a rare cause of sideroblastic anemia.
Polycythemias, or elevated red cell mass, represent a group of erythroid disorders due to nonneoplastic and neoplastic etiologies. Clinical manifestations are typically related to underlying cause rather than the erythrocytosis itself.
DISORDERS OF WHITE BLOOD CELLS
Group of neutrophil disorders, due to nonneoplastic and neoplastic etiologies, resulting in neutropenia.
CASE STUDY: Secondary Polycythemia Due to Hypoxia
Secondary polycythemia is a non-neoplastic condition of increased red blood cell mass.
Secondary polycythemia refers to the increased red blood cell mass not related to a primary erythroid neoplasm.
A 55-year-old female with a long-standing history of chronic obstructive pulmonary disease and sleep apnea presents to her primary physician for routine follow-up. She has been a heavy smoker for approximately 15 years.
CBC Data
WBC count: 10,000/μL
Hgb: 15.8 g/dL
MCV: 93
Platelet count: 400,000/μL
The morphologic features of the PB smear confirm increased numbers of red cells, but are otherwise unremarkable.
In this clinical context, the cause of the increased Hgb concentration is likely the result of increased erythropoietin production as a consequence of hypoxia. A similar mechanism is also responsible for altitude-associated polycythemia. Other secondary causes of increased red cell mass include paraneoplastic syndromes, hereditary disorders of Hgb and erythropoietin homeostasis, and rare hemoglobinopathies.
CASE STUDY: Polycythemia Vera
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by red blood cell production independent of the mechanisms that normally regulate erythropoiesis.
A 54-year-old woman is referred to a hematologist for a several month long history of progressive headaches and erythromelalgia of the lower extremities. Physical examination reveals mild splenomegaly and facial plethora.
CBC Data
WBC count: 15,000/μL
Hgb: 18.5 g/dL
Platelet count: 500,000/μL
Other Laboratory Findings
Erythropoietin: 3 (ref range: 4–27 mU/mL)
PCR for V617F JAK2 mutation: POSITIVE
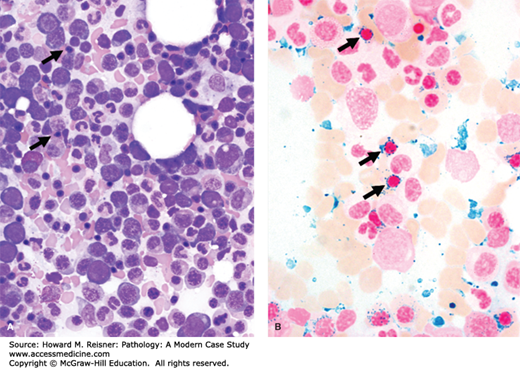
PV is a chronic myeloproliferative neoplasm characterized by red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The identification of the acquired activating mutation of the JAK2 kinase (V617F) is identified in nearly all cases of PV, and along with decreased erythropoietin levels, supports this diagnosis. The BM (Figure 14-3) is typically hypercellular with expansion of all myeloid lineages. The erythroid morphology is normal. Hyperlobated and increased megakaryocytes are commonly identified, sharing morphologic features with other myeloproliferative neoplasms. The clinical course is generally indolent, but the risk of transformation to MDS or acute leukemia is not insignificant.
FIGURE 14-3
PV. The hematoxylin and eosin (H&E)-stained BM biopsy sections demonstrate a markedly hypercellular BM with expansion of the erythroid series (arrows) and appropriate granulopoiesis (×400). Numerous megakaryocytes are also present (arrow heads), although this feature does not effectively distinguish PV from some other myeloproliferative neoplasms.
An absolute neutrophil count (ANC) <1500/μL (<1.5 × 10(9)/L) is the generally accepted definition of neutropenia, as well as the threshold for neutrophil toxicity and infectious risk following chemotherapy. The various causes of neutropenia are listed in Table 14-3. Neutropenia may be acquired or congenital. Acquired causes are much more common than congenital causes. Many drugs can cause agranulocytosis and neutropenia. About three-fourths of all agranulocytosis in the United States is related to drugs. The mechanism of neutropenia varies, depending on the drug. Many antineoplastic drugs cause agranulocytosis and neutropenia by BM suppression. Neutropenia and agranulocytosis can also result from antibody or complement-mediated damage to the stem cells. Some drugs may cause increased peripheral destruction of white cells. Procainamide, antithyroid drugs, and sulfasalazine are at the top of the list of drugs causing this problem. Most agranulocytosis is related to the direct effect related to its dose. Phenothiazines, semisynthetic penicillins, nonsteroidal anti-inflammatory drugs (NSAIDs), aminopyrine derivatives, benzodiazepines, barbiturate, gold compounds, sulfonamides, and antithyroid medications are the most common causes of neutropenia and agranulocytosis. The neutropenia manifests in about 1–2 weeks after exposure to these drugs. Degree of neutropenia depends upon the dose and duration of exposure. Recovery usually occurs within few days of stopping the drug. The marrow recovery may take 10–14 days. Sometimes, a rebound leukocytosis may occur. If the neutropenia is not very severe and the medication is an essential drug for the patient, the drug may be continued with under close monitoring. As long as ANC is above 500–700 and there is no active infection, the drug may be continued if needed. Some drugs may cause increased peripheral destruction of white cells. Neutropenia and agranulocytosis can also result from antibody or compliment-mediated damage to the stem cells. Neutropenia may also be immune mediated in association with an autoimmune disorder or secondary to a drug-induced neutropenia.
|
CASE STUDY: Neutropenia Due to Drug Therapy
Neutropenia may occur secondary to drug therapy and the most common drug-induced blood dyscrasia is a neutropenia.
A 52-year-old male with a kidney transplant a year previously presents with leukopenia. Medications include alemtuzumab, valganciclovir, trimethoprim, sulfamethoxazole, and mycophenolate mofetil. The patient has been undergoing plasmapheresis for recurrent membranous nephropathy.
CBC Data
WBC count: 700/μL
Hgb: 10.3 g/dL
Platelet count: 299,000/μL
ANC: 400/μL
The mycophenolate mofetil is decreased in dosage, and within 2 months the WBC count increases to 6100/μL with an ANC of 5200/μL. In addition, the Hgb level increases to 13.1 g/dL. This approach supports the most likely drug-induced neutropenia.
Other causes of acquired neutropenia should also be considered, including a possible primary BM disorder, particularly a MDS, especially in light of the associated anemia.
Infections may also cause neutropenia. Although most bacterial infections stimulate an increase in neutrophils, some bacterial infections such as typhoid fever and brucellosis and many viral diseases, including hepatitis, influenza, rubella, rubeola, and mumps, decrease the neutrophil count. An overwhelming infection can also deplete the BM of neutrophils and produce neutropenia.
Neutropenia may occur alone or in association with anemia and/or thrombocytopenia in a MDS (i.e., refractory neutropenia or refractory cytopenia with multilineage dysplasia). As mentioned previously, acquired causes of neutropenia are much more common than congenital causes. Refer again to Table 14-3 for the list of congenital causes of neutropenia.
HUNTING FOR ZEBRAS Cyclic Neutropenia
Cyclic neutropenia is a rare (1–2 per million,) typically autosomal-dominant inherited disorder with variable expression, usually presenting in the first year of life.
It is characterized by neutropenia that recurs every 14–35 days, although over 90% of patients exhibit a cycle period of 21 days. While the disease tends to be benign, several affected patients have died of infection. The disorder has been found to be due to germ line mutations in ELA2. ELA2 encodes neutrophil elastase. Interestingly, molecular studies have demonstrated that this protein represents an oncoprotein. Kostmann syndrome is also associated with ELA2 mutations as well as granulocyte colony-stimulating factor receptor (GCSF-r) mutations, and approximately 20% will go on to develop MDS and/or AML.
Group of neutrophil disorders, due to nonneoplastic and neoplastic etiologies, resulting in neutrophilia.
Neutrophilia is defined as an increase in the ANC, which may vary depending on the normal range within the testing laboratory. Various causes of neutrophilia are listed in Table 14-4. Secondary causes of neutrophilia are much more common than primary causes. A true increase in neutrophil production most often reflects an infection, particularly an acute bacterial infection. Neutrophilia can occur from acute infections caused by cocci (e.g., staphylococci, pneumococci, streptococci, meningococci, and gonococci), bacilli (i.e., Escherichia coli, Pseudomonas aeruginosa, and Actinomyces species), certain fungi (i.e., Coccidioides immitis), spirochetes, viruses (i.e., rabies, poliomyelitis, herpes zoster, smallpox, and varicella), rickettsia, and parasites (i.e., liver fluke). Neutrophilia may also be seen with furuncles, abscesses, tonsillitis, appendicitis, otitis media, osteomyelitis, arthritis, cholecystitis, salpingitis, meningitis, diphtheria, plague, and peritonitis. Refer to Table 14-4 for the list of other secondary causes of neutrophilia.
|
Although secondary causes of neutrophilia are much more common than primary causes, a primary neutrophilia should be excluded. Neutrophilia may represent the initial finding in myeloproliferative neoplasms, particularly chronic myelogenous leukemia (CML).
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm characterized by an initial major finding of neutrophilia. CML originates in an abnormal pluripotent BM stem cell and is consistently associated with the BCR–ABL1 fusion gene located in the Philadelphia chromosome, which is found in all myeloid lineages.
Rarely, an absolute neutrophilia may also be associated with nonhematopoietic tumors (i.e., squamous cell carcinoma) as a paraneoplastic phenomenon secondary to cytokine production by the tumor.
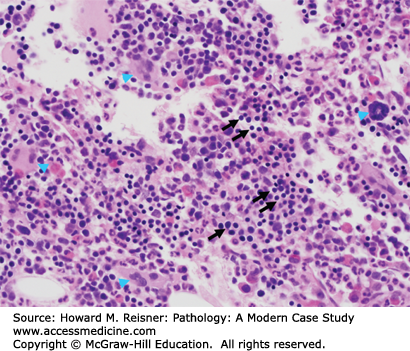
CASE STUDY: Neutrophilia Due to Arthritis
A 4-year-old male presents with 7–10-day history of high spiking fever, rash, and aching in the wrists and knees.
CBC Data
WBC count: 36,400/μL
Hgb: 10.6 g/dL
Platelet count: 662,000/μL
ANC: 32,100/μL (Figure 14-4)
He was diagnosed with juvenile idiopathic arthritis and begun on methotrexate, anakinra (IL-1 receptor antagonist), and prednisone (20 mg b.i.d. for total 40 mg daily). A follow-up visit 5 weeks after initial presentation revealed resolution of symptoms and a WBC count of 8800/μL with and ANC of 7000/μL.
In adults, an absolute lymphocytosis is defined as an increase in the peripheral blood lymphocyte count (ALC) of greater than 4000/μL; in older children (up to 6 years of age), greater than 7000/μL; and in infants, greater than 9000/μL. A relative lymphocytosis occurs when there is a higher proportion (greater than 40%) of lymphocytes among the white blood cells (WBCs); however, the ALC is normal (less than 4,000/μL). A relative lymphocytosis is normal in children under the age of 2 years.
HUNTING FOR ZEBRAS Paraneoplastic Neutrophilia
Paraneoplastic leukemoid reaction is caused by nonhematopoietic tumor-producing hematopoietic growth factors, including GCSF (or other hematopoietic cytokines, such as IL-6), without BM involvement.
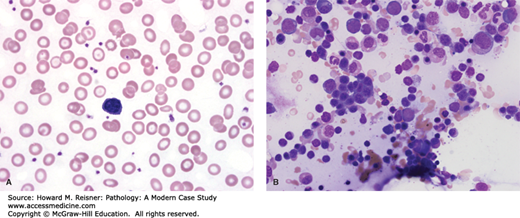
CASE STUDY: Chronic Myelogenous Leukemia
A 40-year-old male presents with a new onset of a diarrheal illness. He presented to a local physician and was noted on physical examination to have an enlarged spleen.
CBC Data
WBC count: 143,900/μL
Hgb: 11.9 g/dL
Platelet count: 589,000/μL
ANC: 122,300/μL
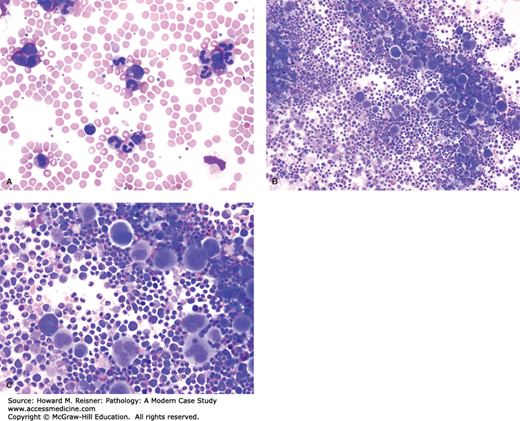
The PB smear reveals a marked leukocytosis with neutrophilia, a few myelocytes and promyelocytes, rare blasts, and a basophilia (4,300/μL) (Figure 14-5A). The BM aspiration and biopsy reveal a markedly hypercellular BM (99% cellular) with small megakaryocytic forms and a markedly increased M:E ratio of 29:1 (Figure 14-5B and C).
Cytogenetic analysis reveals a t(9;22)(q34;q11.2) and DNA studies reveal BCR–ABL p210 transcripts.
FIGURE 14-5
Chronic myelogenous leukemia. The PB smear (A, Wright stain, ×600) demonstrates an increase in neutrophils and granulocytic precursors. An occasional cell has basophilic granules within the cytoplasm. The BM aspirate (B and C, Wright stain; B- ×400; C- ×600) reveals a markedly hypercellular marrow with increased megakaryocytes (with clustering and numerous small forms), a predominance of granulocytic cells, markedly decreased erythroid elements, and scattered eosinophils and mast cells.
An absolute lymphocytosis in children and young adults most often reflects an infection, particularly a viral infection (i.e., infectious mononucleosis) or an infection with pertussis. In older adults with an absolute lymphocytosis, a chronic lymphoproliferative disorder should be considered and excluded. Various causes of an absolute lymphocytosis are listed in Table 14-5.
|
Lymphocytoses may be due to various reactive causes, including viral agents, as well as due to clonal proliferations, and are composed of mature lymphocytes.
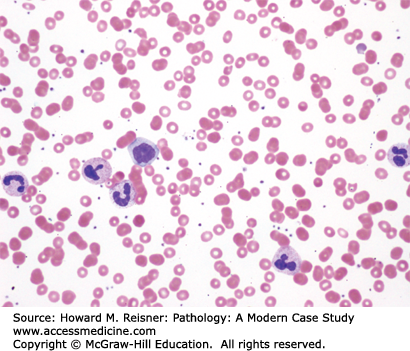
CASE STUDY: Infectious Mononucleosis
Infectious mononucleosis is due to a viral infection with Epstein–Barr virus (EBV), resulting in a reactive peripheral lymphocytosis, composed predominantly of T cells.
A 22-year-old college student presents with a recent history of sore throat, fever, and palpable cervical lymphadenopathy and splenomegaly.
CBC Data
WBC count: 63,600/μL
Hgb: 9.9 g/dL
Platelet count: 256,000/μL
ALC: 45,300/μL
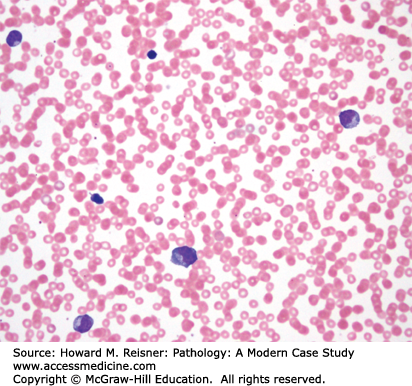
Review of the PB smear reveals a marked increase in atypical lymphocytes (Figure 14-6). Flow cytometric analysis of the PB reveals 78% of cells within the lymphocyte region. Cells within the lymphocyte region are composed of 87% T cells (CD4:CD8 ratio is 0.7) without an aberrant immunophenotype, 8% polyclonal B cells, and 5% natural killer (NK) cells. The patient subsequently had a positive Mono spot test. The patient was diagnosed with infectious mononucleosis.
The most common chronic lymphoproliferative disorder presenting with an absolute lymphocytosis is chronic lymphocytic leukemia (CLL). CLL is also the most common leukemia of adults in Western countries.
The morphologic features of CLL may also be confused with the malignant cells composing the small cell variant of T-cell prolymphocytic leukemia (T-PLL), which has a much more aggressive clinical course.
T-PLL is an aggressive T-cell leukemia characterized by the proliferation of small-to-medium-sized prolymphocytes with a mature post-thymic T-cell phenotype involving the PB, BM, lymph nodes, spleen, and skin. The morphology of the leukemic cells may range from those of typical prolymphocytes with a single prominent nucleolus to small forms with no apparent nucleolus (i.e., the small cell variant), which may mimic the cells of CLL.
T-PLL is rare, representing approximately 2% of cases of mature lymphocytic leukemias. It is important to distinguish it from CLL, as described above, since T-PLL has a much more aggressive clinical course and is treated differently.
Monocytopenia is relatively rare and defined as an abnormally low level of monocytes in the PB (i.e., less than 200/μL) and can be seen in non-neoplastic (i.e., HIV infection) and neoplastic (i.e., hairy cell leukemia) disorders.
CASE STUDY: Chronic Lymphocytic Leukemia
CLL is a neoplasm composed of monomorphic and clonal small, round B lymphocytes. The cells usually coexpress CD5 and CD23. In the absence of extramedullary tissue involvement, there must be greater than 5000/μL peripheral monoclonal lymphocytes with a CLL phenotype.
A 49-year-old male is referred for the evaluation of possible CLL. He presented to his primary care provider with mild fatigue, but few other symptoms. A routine CBC showed a white count of 120,000 that were predominantly lymphocytes.
Upon presentation at referral, his CBC data is as follows:
WBC count: 149,500/μL
Hgb: 15.1g/dL
Platelet count: 119,000/μL
ALC: 123,200/μL
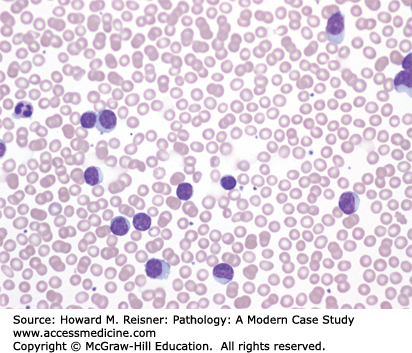
Review of the PB smear reveals a marked increase in the number of small, round, mature lymphocytes (Figure 14-7). Flow cytometric analysis of the PB reveals 92% cells within the lymphocyte region. Cells within this region are composed of 2% T cells (CD4:CD8 ratio is 1:1) without an aberrant immunophenotype, 96% monoclonal B cells, and 2% presumed NK cells. The monoclonal B-cells variably express CD19, CD20, CD23, CD79b, CD38, selective kappa surface light chains, aberrant CD5, and ZAP-70. They do not express CD10. This immunophenotype is characteristic of CLL, representing 88% of the peripheral WBCs. ZAP-70 expression is associated with a worse prognosis in CLL. Cytogenetic analysis reveals an abnormal karyotype: 46,XY,del(11)(q?14q?23),del(13)(q?12q?14)[1]/46,XY[4]. Del (13) is the most common abnormality identified in CLL by FISH analysis.
CASE STUDY: T-PLL HUNTING FOR ZEBRAS
A 66-year-old male with a long-standing outside history of “chronic lymphocytic leukemia” (no chemotherapy to date) presents to an outside hospital with bloody diarrhea and hematemesis and is noted to have a markedly elevated WBC count. He is then transferred to a major medical center for further care.
Upon presentation at the major medical center, CBC data is as follows:
WBC count: 117,100/μL
Hgb: 9.2 g/dL
Platelet count: 45,000/μL
ALC: 90,400/μL
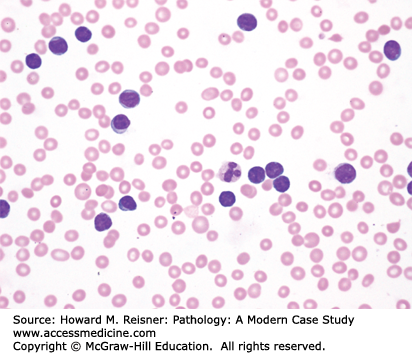
Review of the PB smear reveals a marked increase in lymphoid forms with variably irregular nuclear contours (Figure 14-8); prolymphocytes are not identified. Flow cytometric analysis of the PB reveals 90% of cells are within the lymphocyte region. Cells within this region are composed of 99% T cells (CD4:CD8 ratio is 1:1) with an aberrant immunophenotype and <1% polyclonal B cells. The aberrant T cells uniformly express CD2, CD3, CD4 and CD8, CD5, CD7, and CD25, and lack expression of CD10, CD1a, CD34, CD56, CD117, and myeloid or B-cell antigens. This immunophenotype is characteristic of a mature aberrant T-cell immunophenotype, representing 90% of the peripheral WBCs and consistent with the small cell variant of T-PLL.
Such a presentation of a reported history of CLL is not uncommon in this variant of T-PLL, due to the overlapping morphologic features with CLL. Such a case supports the importance of performing flow cytometric immunophenotyping in all cases suspected of “CLL.”
CASE STUDY: Monocytopenia in AIDS
A 48-year-old male with a history of HIV/AIDS presents with a 2-month history of progressive cough and fever. He also has a warm autoimmune hemolytic anemia (receiving weekly rituximab), resolving candidemia, splenomegaly, and worsening thrombocytopenia.
CBC Data
WBC count: 4600/μL
Hgb: 6.6 g/dL
Platelet count: 18,000/μL
ANC: 4100/μL
ALC: 400/μL (normal: 2000–4000/μL)
Monocyte count: 0.
Monocytopenia typically also occurs in hairy cell leukemia, and should be considered in patients presenting with unexplained pancytopenia in the appropriate clinical context.
An absolute monocytosis is defined as a peripheral monocyte count greater than 1000/μL. The various causes of an absolute monocytosis are listed in Table 14-4.
Monocytoses may be due to various reactive causes as well as to clonal disorders. Reactive causes of an absolute monocytosis should be sought, particularly in adult patients. In an adult patient, an unexplained, persistent (greater than 3 months) absolute monocytosis should be considered a clonal process until proven otherwise.
CASE STUDY: Monocytosis Due to Bacterial Infection
Postoperative states may be associated with a peripheral monocytosis.
A 83-year-old female with a history of fracture status-post hip replacement presents with leukocytosis and anemia post surgery.
CBC Data
WBC count: 52,900/μL
Hgb: 10.8 g/dL
Platelet count: 377,000/μL
NEUT: 34,700/μL
Monocyte count: 9200/μL
Two months after presentation, the monocytosis had resolved. Thus, this original finding was attributed to postoperative infection. Unless proven otherwise, a persistent absolute monocytosis in an adult should be considered a clonal/neoplastic process.
Chronic myelomonocytic leukemia (CMML) is a neoplasm composed of monocytic cells at various stages of differentiation.
CMML is a clonal BM stem cell disorder in which monocytosis is a major defining feature. Diagnostic criteria for CMML are as follows:
Persistent PB monocytosis (>1000/μL)
No Philadelphia chromosome or BCR–ABL fusion gene
Fewer than 20% blasts (including myeloblasts or monoblasts) and/or blast equivalents (promonocytes: monocytic cells with a single prominent nucleolus) in the PB or BM
Dysplasia in one or more myeloid cell lines.
If myelodysplasia is absent or minimal, the diagnosis of CMML may still be made if the other requirements are met (i.e., an acquired, clonal cytogenetic abnormality is detectable in the BM cells or the monocytosis has persisted for 3 months or longer, and all other causes of monocytosis have been excluded).
If greater than 20% blasts and/or promonocytes are present in the PB or BM, the World Health Organization (WHO) classification diagnosis is AML.
CMML is further divided into CMML-1 and CMML-2, based on the percentage of blasts and/or promonocytes:
In CMML-1, blasts and/or promonocytes represent fewer than 5% of peripheral WBCs (WBCs) and fewer than 10% of BM nucleated cells.
In CMML-2, blasts and/or promonocytes represent between 5% and 19% of peripheral WBCs or between 10% and 19% of BM cells. CMML-2 may also be diagnosed when there are fewer than 20% blasts in the PB or BM if Auer rods are identified.
It should always be kept in mind that a diagnosis of CMML should never be based on PB findings only and without BM examination, since there may be a higher percentage of immature cells (blasts and/or promonocytes) in the BM, meeting the criteria for AML.
A persistent, absolute monocytosis may also be associated with lymphomas and more rarely carcinomas as a paraneoplastic phenomenon. Paraneoplastic monocytosis is caused by nonhematopoietic tumor-producing hematopoietic growth factors, including GM-CSF, without BM involvement.
DISORDERS OF PLATELETS
Group of platelet disorders, due to non-neoplastic and neoplastic etiologies, leading to thrombocytopenia.
Platelets are an integral part of the coagulation system and hemostasis. Their function is complex, and they serve to form a mechanical plug at sites of vascular injury, and as important reservoirs of other procoagulant factors. A variety of disorders are responsible for decreased numbers of platelets, or thrombocytopenia, but qualitative defects of platelet function have similar sequelae. Table 14-6 highlights both quantitative and qualitative platelet disorders resulting in thrombocytopenia. Clinical manifestations of thrombocytopenia typically include bleeding from mucosal sites and petechial hemorrhages on the skin.
| Quantitative | Qualitative | |
|---|---|---|
| Decreased production | Destruction or sequestration | Congenital |
| Congenital | Splenomegaly | Bernard–Soulier syndrome |
| B12/folate deficiency | ITP | Glanzmann thrombasthenia |
| Aplastic anemia | DIC | Acquired |
| Myelodysplastic syndrome | Other autoimmune disease | Aspirin |
| Mass effect | Uremia | |
| Bone marrow metastasis | Liver failure | |
| Acute leukemia | ||
| Myelofibrosis | ||
CASE STUDY: CMML
This case demonstrates that although the “clonal” monocytic cells in CMML may appear mature morphologically, a combination of ancillary studies (FCI, and/or cytogenetic and molecular analyses) aids in distinguishing reactive from clonal processes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


