TABLE 12-1 PHASES OF INTRAUTERINE LUNG DEVELOPMENT | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
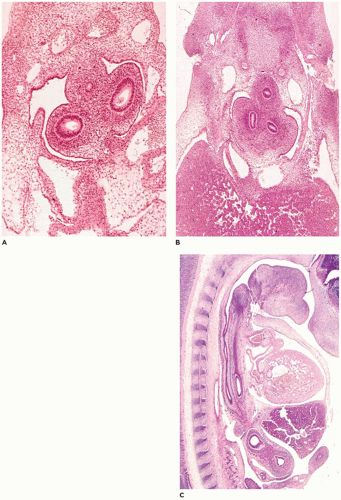 FIGURE 12-1 • Embryonic periods in respiratory tract development. A: At 29 to 31 days gestation (stage 14), the primary bronchial buds are surrounded by primitive mesenchyme. Note the small esophagus above and between the bronchi. B: By 35 to 37 days (stage 16), the primary bronchi have divided into secondary and early tertiary buds. Note the centrally located esophagus and the large amount of hepatic parenchyma (lower half). C: In a sagittal plane of a 37- to 40-day (stage 17) embryo, the relationship between the esophagus (nearest vertebral column) and trachea (between esophagus and heart) can be seen. The heart and liver are ventral to the foregut structures. |
Type I pneumocytes then differentiate from type II cells to form the thin air-blood interface required for gas exchange. As the interstitium thins in the latter portion of the acinar period, the capillaries of the interstitium proliferate and come to lie beneath the type I cells. Submucosal glands in the trachea and bronchi progress from tubules to mucuscontaining acini. By week 24, the cartilage has extended to the most distal bronchi.
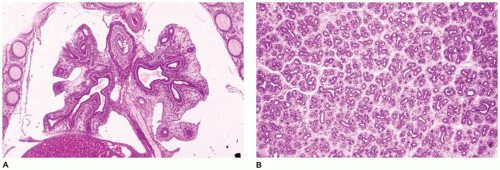 FIGURE 12-2 • Pseudoglandular period. A: At 9 weeks of gestation, the proximal airways are present throughout the right and left lobes. B: By 13 weeks, bronchiolar development is well under way, and early division into lobules and clusters of acini is apparent. |
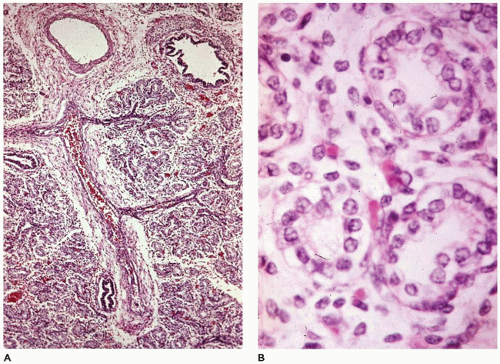 FIGURE 12-3 • Acinar period. A: In a 370-g fetus, acinar development is characterized by pulmonary arteries and proximal bronchioles surrounded by alveolar ducts still widely separated by mesenchymal tissue. B: The alveolar duct structures are lined by cuboidal epithelium (early type II cells), and blood-filled capillaries are present just beneath the cells. |
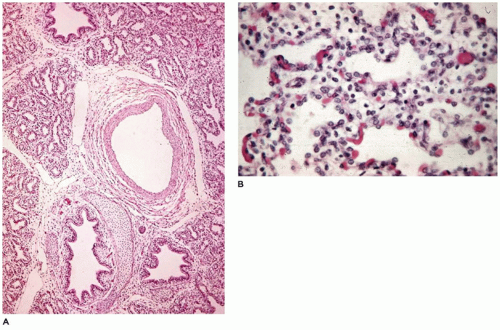 FIGURE 12-4 • Saccular period. A: In a 650-g fetus, discrete acini are identifiable within a lobule. B: Secondary crests are covered by thinning type I cells, which expose capillary beds immediately beneath the cells. |
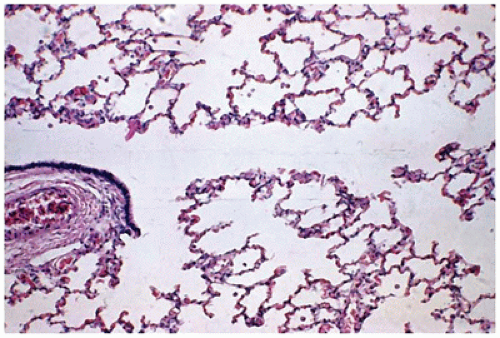 FIGURE 12-5 • Alveolar period. At 2 months of age, a respiratory bronchiole (left) gives rise to alveolar ducts, alveolar saccules, and thin-walled alveoli. |
recessive form. It has been associated with palatal defects, tracheoesophageal fistula (TEF), congenital heart malformations, trisomy 6, Pfeiffer syndrome, Treacher Collins syndrome, the fetal carbimazole syndrome, and the CHARGE (Coloboma, Heart defect, choanal Atresia, Retardation, Genital, Ear anomaly) association (CHD7 mutation on 8q12.2) of which it is a major component.
Type 1, atresia of both supraglottic and infraglottic portions of the larynx
Type 2, atresia of the infraglottic region (Figure 12-6A, B)
Type 3, glottic atresia
Supraglottic interarytenoid cleft (50% of cases)
Partial cricoid cleft
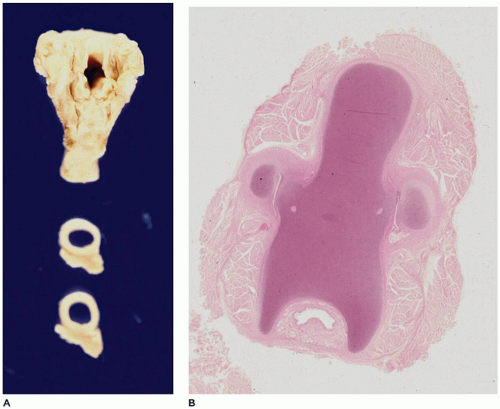
FIGURE 12-6 • Laryngeal atresia. A: The larynx reveals a patent upper opening (upper piece) and a patent trachea (lower two cross sections). B: A histologic section from the area in the region of the cricoid cartilage reveals only a pinpoint lumen (bottom center).
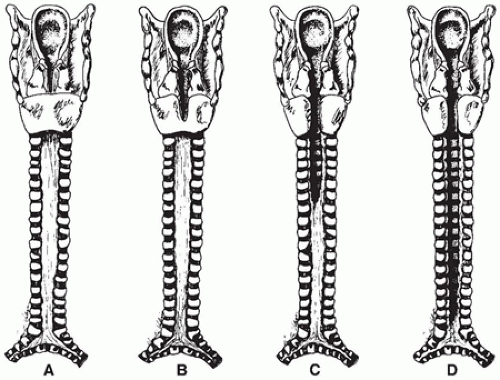
FIGURE 12-7 • Types of laryngotracheoesophageal cleft. A: Supraglottic interarytenoid cleft. B: Partial cricoid cleft. C: Total cricoid cleft. D: Complete cleft to level of carina.
Total cricoid cleft
Complete cleft of the trachea to the level of the carina
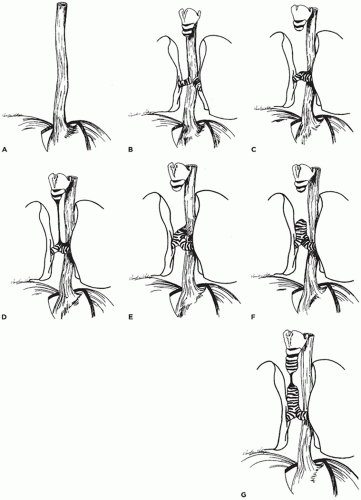 FIGURE 12-8 • Types of tracheal agenesis. A: Total pulmonary agenesis (8% of cases). B: Tracheal agenesis with main bronchi arising directly from the esophagus (10%). C: Tracheal agenesis with fused main bronchi and bronchoesophageal fistula (56%). D: Tracheal agenesis with the larynx joined by atretic strand to distal trachea, which has a fistulous connection with the esophagus (10%). E: Upper tracheal agenesis with large direct tracheoesophageal communication (5%). F: Tracheal agenesis with no communication with the esophagus (5%). G: Short-segment tracheal agenesis (5%). |
however, nearly 70% of cases consist of agenesis of the entire trachea with a small fistulous connection between the esophagus and the carina (Figure 12-8C to E) (23,24). The lungs may be normally developed or totally absent (pulmonary agenesis). In the rare cases of tracheal agenesis with no fistulous connection to the esophagus (i.e., total sequestration of the lungs), the lungs are uniformly distended, histologically resembling extralobar sequestration (25). There is a male predominance of approximately 2:1 and an association with maternal polyhydramnios in tracheal agenesis. In addition to the anomalies of the VATER association, tracheal agenesis has been seen in association with duodenal atresia, annular pancreas, syndactyly, and CNS malformations. Evans et al. (24) describe four groups based on the type of anomalies associated with the tracheal agenesis: group 1, anomalies restricted to the trachea, larynx, and cardiovascular system; group 2, severe cardiovascular anomalies and abnormal lung lobulation; group 3, a caudal component in addition to thoracic abnormalities, with anal and renal anomalies being common; and group 4, multisystem involvement with a high incidence of aberrant vessels, complex cardiac malformations, lung lobation defects, and anomalies of other foregut derivatives.
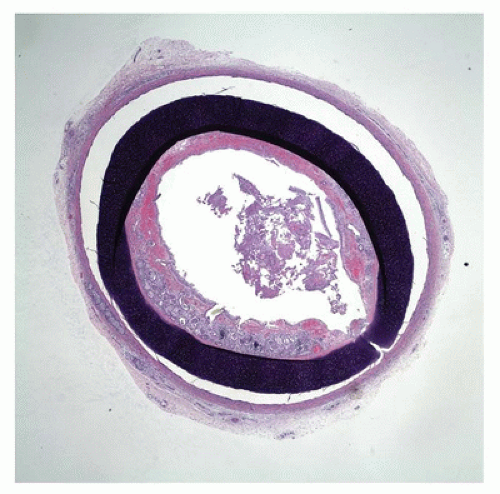 FIGURE 12-9 • Tracheal stenosis. A cross section from the mid trachea shows a complete cartilage ring beneath the mucosa, significantly narrowing the tracheal lumen. |
is present in more than 30% of cases, and nearly 35% of the infants are premature. The anomaly can be divided into five (or more) types (Figure 12-10A to E). More than 95% of the patients have EA with the clinical findings of excessive oral and pharyngeal secretions or choking, cyanosis, or coughing during first attempts at feeding. While 98% are sporadic, the remaining have separate genetic factors (34).
 FIGURE 12-10 • Types of tracheoesophageal fistula (TEF) and esophageal atresia (EA). A: EA with TEF to the distal esophageal segment (>85% of cases in various series). B: EA without TEF (8%). C: TEF without EA (4%). D: EA with TEF to the proximal esophageal segment (1%). E: EA with TEF to both proximal and distal esophageal segments (1%). |
and intracardiac epithelial cyst. TEFs may develop in burn patients, with foreign body impaction such as a disc battery (36), and following radiation and chemotherapy for mediastinal malignancies, including lymphoma (37).
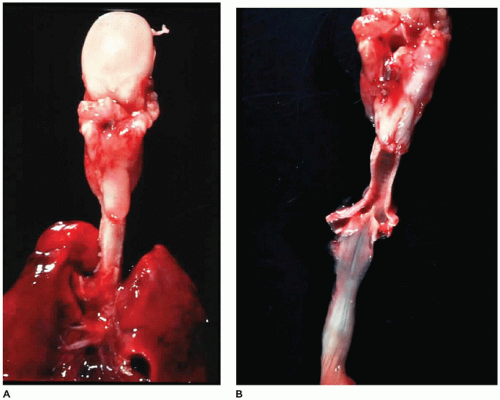 FIGURE 12-11 • TEF and EA. A: In a posterior view of the tongue (top), trachea, and lung, the esophagus is seen to end in a blind pouch (center). B: With the trachea and esophagus open posteriorly, a fistula can be seen connecting the carina with the distal end of the esophagus. |
TABLE 12-2 ANOMALIES ASSOCIATED WITH ESOPHAGEAL ATRESIA AND TRACHEOESOPHAGEAL FISTULA | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
and the bronchi supplying the right middle and lower lobes are branches of the left main bronchus that are crossing or “bridging” the mediastinum (Figure 12-13). They note that the origin of the tracheal bronchus is at the normal level of tracheal bifurcation (T4-5), and the bifurcation of the bronchi supplying the left lung and right middle and lower lobes is at the T6-7 level.
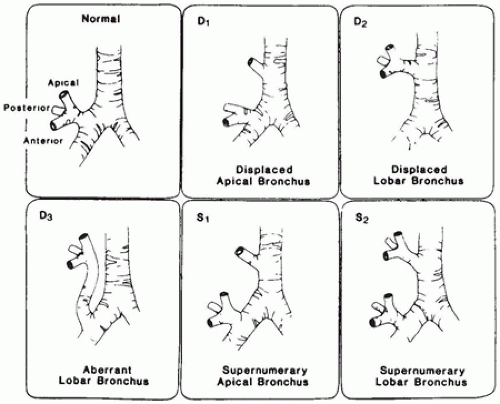 FIGURE 12-12 • Anatomic variations of right upper lobe bronchus. (From McLaughlin FJ, Strieder DJ, Harris GB, et al. Tracheal bronchus: association with respiratory morbidity in childhood. J Pediatr 1985;106:751, with permission.) |
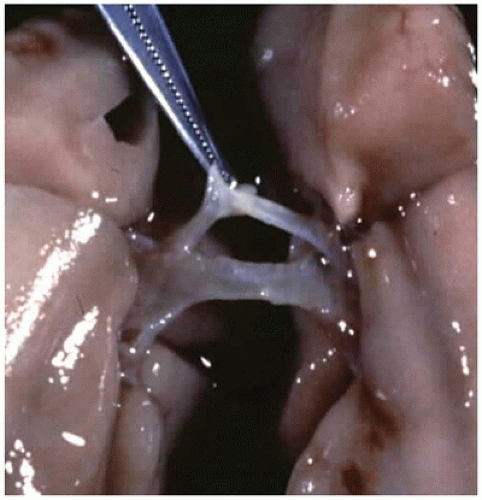 FIGURE 12-13 • A bronchus “bridges” the mediastinum in the case of tracheal agenesis. |
reports of “intrapulmonary bronchogenic cysts” probably represent instances of type 1 CPAM (52).
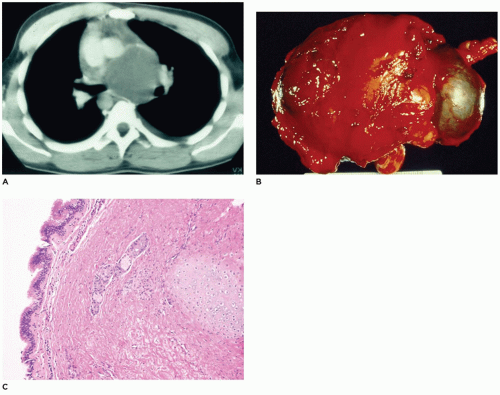 FIGURE 12-14 • Bronchogenic cyst. A: A CT of the chest displays a large mass in the middle mediastinum. B: A resected bronchogenic cyst, which was separate from the lung, is covered by connective tissue. C: Ciliated pseudostratified columnar epithelium overlies a wall composed of fibrous connective tissue, glands, and a cartilage plate in a bronchogenic cyst. |
of mucin. Other underlying causes include cystic fibrosis (CF), neoplasia, alpha-thalassemia, beta-thalassemia, and acute chest syndrome of sickle cell disease. Secretory hyperresponsiveness with excess mucin secretion is regarded as the basis for the disorder (57).
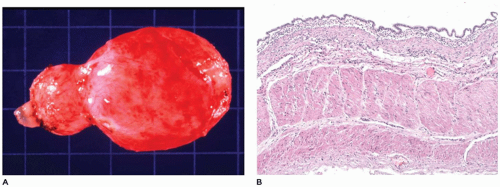 FIGURE 12-15 • Esophageal cyst. A: A cystic structure was resected from the middle mediastinum adjacent to the esophagus. B: Columnar epithelium overlies a wall composed of thick muscular bands in this esophageal cyst from the mediastinum. |
and diaphragmatic defect), among others (65). Bronchial supply to both lungs may be anatomically normal, but there is usually an anomalous pulmonary artery supply and venous drainage resembling that seen in scimitar syndrome.
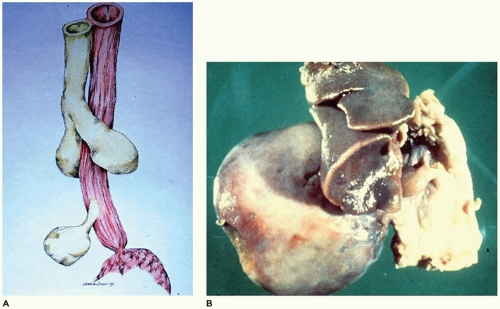 FIGURE 12-16 • Extralobar sequestration. A: The normal lung develops as an evagination from the foregut (top half). A second evagination (bottom) from the foregut gives rise to lung tissue not attached to the normally developing lung. B: A large right-sided thoracic mass is attached to the mediastinum by a thin vascular pedicle. Note the hypoplasia of the right lung. |
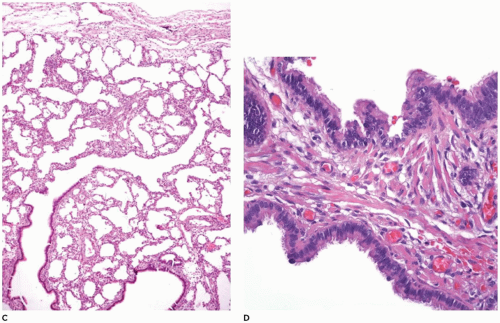 FIGURE 12-16 • (Continued) C: The pulmonary parenchyma is uniformly dilated from the bronchioles to the most distal alveoli. D: Back-to-back bronchiole-like structures typical of CPAM type 2 are seen in 50% of extralobar sequestrations. Note also the rhabdomyomatous dysplasia. |
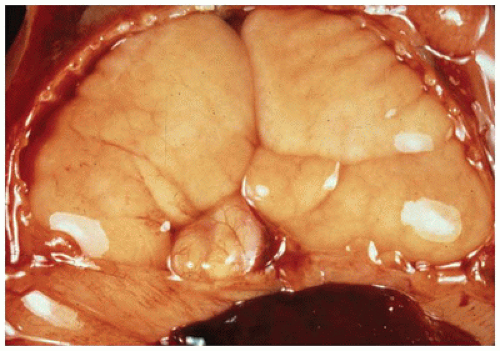 FIGURE 12-17 • Hyperplastic lungs in the case of laryngeal atresia are massively enlarged, displaying the markings of the ribs on their surface. |
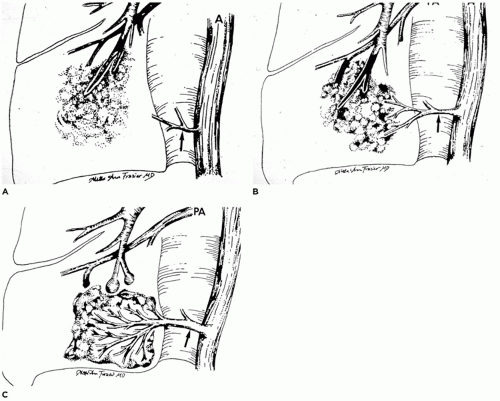 FIGURE 12-18 • Sequence of events in the formation of intralobar pulmonary sequestration. A: Occlusion of a bronchial branch by means such as aspirated material or inflammatory debris can lead to the development of pneumonia distal to the occlusion. B: As the pneumonia persists or progresses, the lung seeks oxygenated blood to aid in resolution and repair. If pulmonary artery flow is inadequate, systemic blood supplying pleural granulation tissue through the pulmonary ligament arteries may be “parasitized.” C: As the pneumonia resolves (or progresses or recurs), the major arterial supply to the sequestered portion of lung is derived from the hypertrophied pulmonary ligament artery (or arteries). (From Stocker JT, Malczak HT. A study of pulmonary ligament arteries: relationship to intralobar pulmonary sequestration. Chest 1984;86:611, with permission.) |
repeated episodes of pneumonia. During the course of these episodes, normal pulmonary ligament arteries become hypertrophic to provide the systemic artery supply (Figure 12-18A to C) (70,72). Some examples of ILS may develop within a previously existing malformation (e.g., CPAM). The following evidence suggests the acquired nature of ILS (66,73):
Microscopically, the pulmonary parenchyma is distorted by chronic inflammation and fibrosis (Figure 12-19D). The cysts are lined with cuboidal or columnar epithelium and are filled with amorphous eosinophilic material, foamy macrophages, or both. Elastic and muscular arteries are present within the interstitium and may show medial hypertrophy, thrombosis, and arteritis (76).
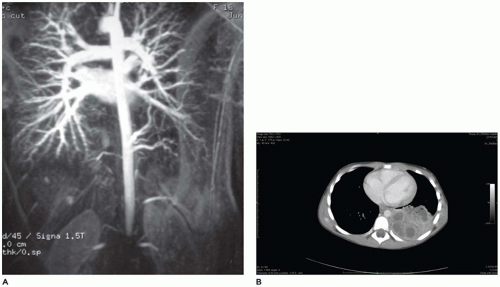 FIGURE 12-19 • Intralobar sequestration. A: An arteriogram demonstrates arteries arising from the descending aorta (mid right) supplying a portion of pulmonary parenchyma. B: A CT demonstrates a mass in the posterior area of the right hemithorax. |
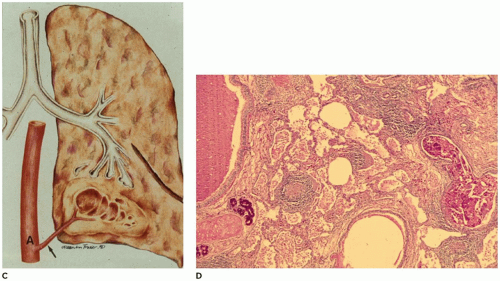 FIGURE 12-19 • (Continued) C: An artery arising from the descending aorta and passing through the pulmonary ligament supplies a cystic portion of the lung in the left lower lobe. D: Dense fibrous connective tissue containing lymphoid aggregates surrounds irregular cysts filled with debris and macrophages. |
the lung (81). As with infants with hypoplasia secondary to other anomalies, these infants present with respiratory distress, are difficult to ventilate, and frequently have episodes of pneumothorax (PT) and interstitial pulmonary emphysema (IPE). Potter sequence with sloping forehead, flattened face and nose, receding chin, large ears, broad spade-like hands, and deformations of the limbs secondary to compression by the uterus in the absence of adequate amniotic fluid is a consistent finding in cases associated with oligohydramnios from any cause. Pulmonary hypoplasia has been noted in children with Down syndrome, but it is thought to result from failure of the lung to develop properly in the postnatal period (83).
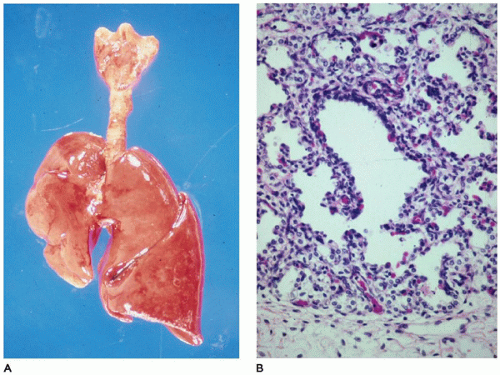 FIGURE 12-20 • Pulmonary hypoplasia. A: The right lung is markedly diminished in size, secondary to herniated abdominal organs through a right-sided diaphragmatic hernia. By weight, the left lung is also hypoplastic. B: At the periphery of an acinus in this hypoplastic lung, a radial alveolar count (RAC) is far below the normal of 4 to 6 for a term infant, confirming the diagnosis of hypoplasia. |
TABLE 12-3 ANOMALIES ASSOCIATED WITH PULMONARY HYPOPLASIA | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
TABLE 12-4 CAUSES OF INFANTILE LOBAR OVEREXPANSION | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Surgical resection of the involved lobe is curative, although nonsurgical management has been successful in unusual cases (85).
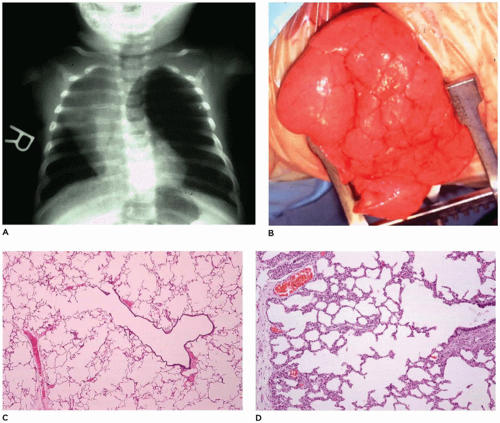 FIGURE 12-21 • Infantile lobar overinflation. A: A hyperinflated left lung shifts the mediastinum to the right. B: At surgery, the hyperinflated lung bulges from the opening in the thorax. C: “Classic” form of ILO. The alveolar duct and alveoli are dilated to 3 to 10 times the normal size but are otherwise unremarkable. D: “Hyperplastic” form of ILO. While not overinflated, this lung displays a complex acinar formation with a larger number of alveoli (and consequently a large radial alveolar count) than would be expected at this age. |
and uniformly dilated and may appear to be increased in number. Identification of lymphatics can be aided by CD31 and D2-40 immunohistochemistry and can help differentiate the lesion from IPE. These small, irregular cysts are lined with a thin layer of endothelial cells and surrounded by a loose myxoid to occasionally dense connective tissue that often contains foci of extramedullary hematopoiesis. Clusters of lymphatics surround bronchovascular bundles within the interlobular septa and may separate acini beneath the pleura (Figure 12-22C, D). This is in contrast to the air-filled, larger, “unlined” cysts of IPE that are limited to the interlobular septa and do not extend laterally beneath the pleura.
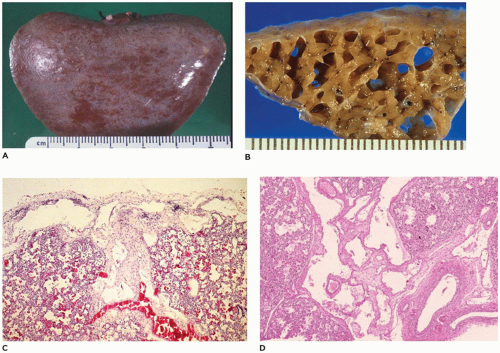 FIGURE 12-22 • Congenital pulmonary lymphangiectasis. A: A fine network of dilated lymphatics is present beneath the pleura, most notably where interlobular septa intersect the pleura. B: Cut section of the lung from an infant total anomalous pulmonary venous return reveals enormously dilated lymphatics. C: Dilated lymphatics extend laterally beneath the pleura (top) and centrally along an interlobular septum (center). Note the slight increase in connective tissue between the channels. D: Numerous dilated lymphatics extend along interlobular septa surrounding bronchovascular bundles. |
surface (Figure 12-24A). Microscopically, tissue consists of bronchus-like structures with muscle, glands, and numerous cartilage plates (Figure 12-24B). Prominent mesenchymal tissue separates these structures and contains extramedullary hematopoiesis, large thin-walled vascular channels, and collections of amorphic basophilic debris. Rarely, structures resembling proximal bronchioles are present, along with a few scattered acini at the periphery of the lesion.
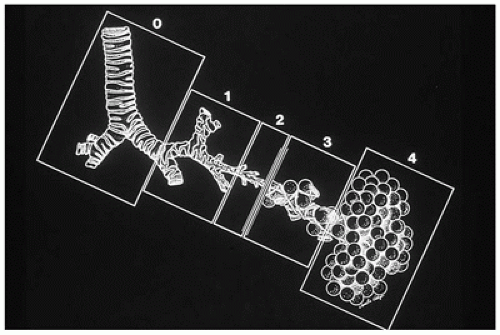 FIGURE 12-23 • Classification of CPAM. The classification is based on the similarity in appearance of the hamartomatous components of the lesion with the various areas of the normal tracheobronchial tree. Type 0, composed of bronchuslike structures, appears to be a malformation of the most proximal tracheobronchial tree. Type 1, containing bronchuslike and proximal bronchiole-like structures, mimics the distal bronchial tree and proximal acinus. Type 2, composed of bronchiole-like structures, resembles the bronchiolar segment of the acinus. Type 3, composed of bronchiole-like structures and alveolar ducts and saccules lined by cuboidal epithelium, resembles the midacinar region. Type 4, with thin-walled structures lined by type 1 alveolar lining cells, should be diagnosed correctly as PPB type 1. (From Stocker JT. Congenital and developmental diseases. In: Dail HD, Hammer SP, eds. Pulmonary pathology. 2nd ed. New York: Springer-Verlag, 1994:182, with permission.) |
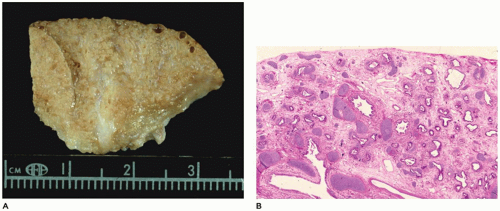 FIGURE 12-24 • CPAM, type 1 (congenital acinar dysplasia). A: A small nodular mass representing the right lung is largely devoid of air. A similar lung was present on the left side. B: Bronchial-like structures are surrounded by irregular cartilage plates and loose mesenchyme-containing thin-walled vascular structures. |
Mucous cells and cartilage plates are absent except as components of “entrapped” normal bronchi. A variant or subgroup of the type 2 lesion, termed rhabdomyomatous dysplasia, contains ribbons of striated muscle fibers throughout the lesion, both in association with the cysts and between alveolar ducts and around blood vessels (Figure 12-27C). The cysts of this rhabdomyomatous variant may be less prominent than other type 2 lesions. Rhabdomyosarcoma has been reported to originate from CPAM, but this likely represents a primary pleuropulmonary blastoma (PPB) rather than being secondary to CPAM. CPAM, type 2-like features are present in 50% of extralobar sequestrations (67).
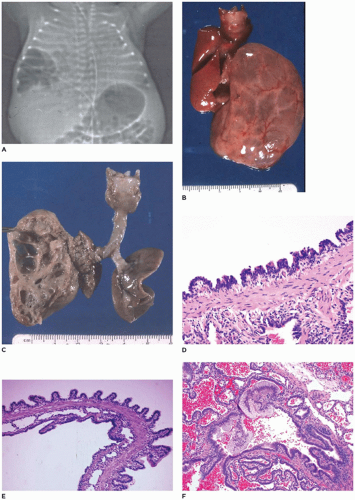 FIGURE 12-25 • CPAM, type 1. A: A cystic mass is present in the lower right hemithorax in a newborn with respiratory distress. B: Multiple large, fluid-filled cysts distend the left lobe from a fetus in the second trimester. C: When opened, the mass consists of intercommunication cysts. D: Cysts of type 1 CPAM are characteristically lined by ciliated intercommunicating in a sawtooth configuration with underlying fibromuscular connective tissue. E: A larger cyst wall (top) is covered by columnar epithelium in a papillary configuration. Note the columnar epithelial lining of the smaller cysts as well. F: Clusters of mucogenic cells are present along the cyst lining. |
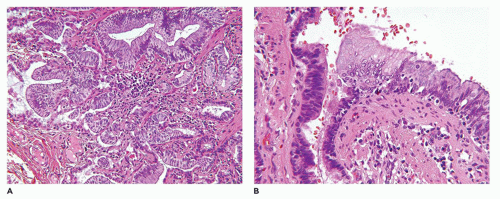 FIGURE 12-26 • CPAM type I. A: Mucinous metaplasia in the lung of an 18-year old who presented with a right middle lobe cyst. B: The mucinous epithelium has low-grade cytologic features, but it is thought that the changes are a precursor to mucinous adenocarcinoma. |
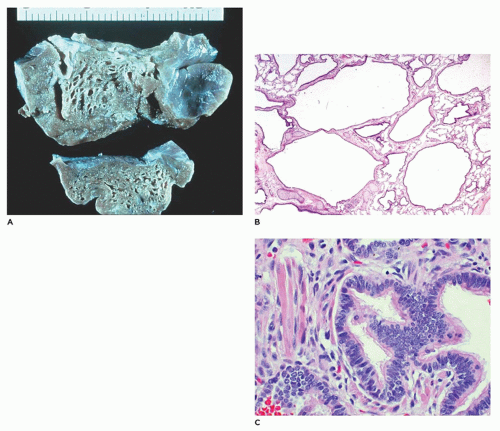 FIGURE 12-27 • CPAM, type 2. A: Small cysts (0.2 to 0.5 cm) are scattered throughout the lobe and blend with normal parenchyma. B: The back-to-back bronchiole-like structures are separated by structures resembling alveolar ducts. C: In a variant of type 2, striated muscle fibers are present in the connective tissue between and around cysts. |
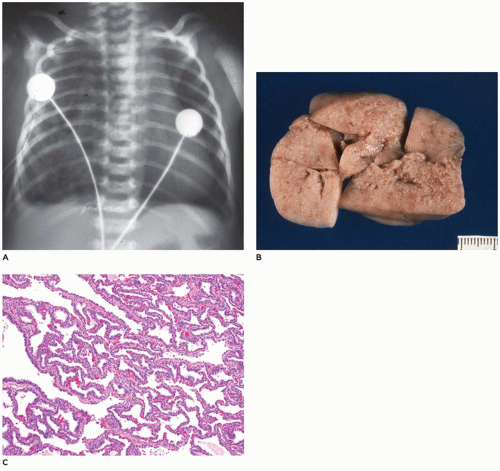 FIGURE 12-28 • CPAM, type 3. A: A large air-containing mass in the right hemithorax pushes the mediastinum to the left. B: The resected lesion is nearly solid with only a few slit-like openings. C: Randomly distributed irregular bronchiole-like structures are separated by dilated alveolus-like structures all of which are lined by cuboidal epithelial cells, imparting an adenomatoid (or gland-like) appearance. |
epithelial cells (type I and II pneumocytes), with occasional low cuboidal epithelium seen (Figure 12-29B to D). The wall of the cyst is composed of loose mesenchymal tissue with prominent arteries and arterioles. It must be emphasized that PPB type 1 and CPAM, type 4 are one in the same and should be viewed and managed as PPB.
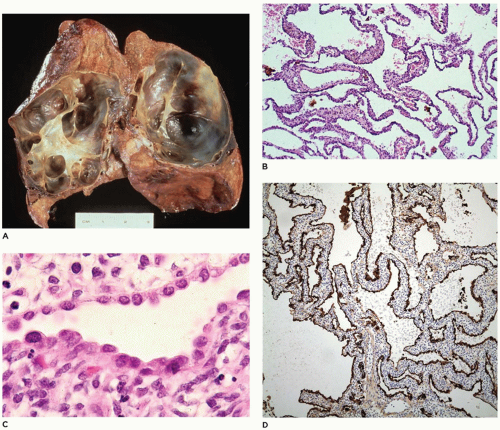 FIGURE 12-29 • PPB, type 1 (in the past diagnosed as CPAM type 4). A: The lung is distended by thin, almost translucent cyst walls. B: The walls of the cysts are composed of loose mesenchyme covered by an indistinct epithelial lining not apparent at this magnification (hematoxylin and eosin stain, original magnification ×25). C: The cyst walls are variously covered by an attenuated epithelium of alveolar lining cells (hematoxylin and eosin stain, original magnification ×150). D: The epithelium stains positively for cytokeratin (H&E, ×50). Note: Because of the similarities between PPB type 1 and CPAM type 4, it is best to consider these lesions as having the potential to progress to PPB types 2 and 3. All patients with such cystic lesions must have complete surgical excision with clear margins. If any doubt exists as to the correct diagnosis, the case should be referred to the International PPB Registry to ensure correct follow-up and management of the patient. |
TABLE 12-5 ANOMALIES ASSOCIATED WITH CONGENITAL CAPILLARY ALVEOLAR DYSPLASIA | |
|---|---|
|
membranes along terminal and respiratory bronchioles and alveolar ducts. In the pediatric age group, DAD has been typically described in neonates with surfactant deficiency, although other causes include infections, inhalational injury, sepsis, and systemic shock. Breathing difficulty is common in the early neonatal period, occurring in up to 7% of newborn infants, and is not synonymous with RDS. Other common causes of breathing difficulty in term newborn infants include transient tachypnea of the newborn, pneumonia, meconium aspiration syndrome (MAS), persistent pulmonary hypertension of the neonate, and PT (111).
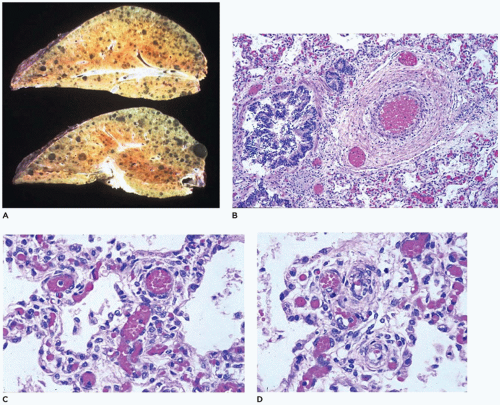 FIGURE 12-30 • Congenital alveolar capillary dysplasia. A: Bulky stiff lungs display focal hemorrhage and prominent interlobular septa. B: Dilated veins are present adjacent to and within the adventitia of a pulmonary artery (center-right). C: Broad alveolar septa contain many centrally located capillaries with only a few of them approaching the alveolar epithelium. D: Muscularized arteries are present within alveolar septa well away from bronchioles. |
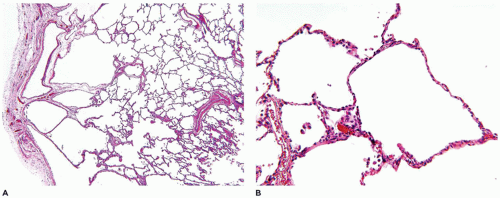 FIGURE 12-31 • Peripheral lung cysts in a 4-year-old male with trisomy 21. A: This lung biopsy demonstrates the presence of multiple pulmonary cysts residing beneath the pleural surface. B: These cysts represent a growth abnormality in the formation of small airspaces. |
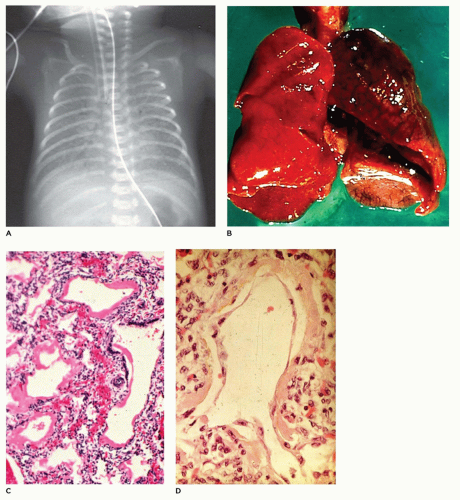 FIGURE 12-32 • Acute lung disease— HMD of the newborn. A: In this 24-hour-old, 1,050-g infant with respiratory distress, the lungs display a classic “ground-glass” opacity. B: The lungs in HMD are often atelectatic and display focal hemorrhage. C: Dilated bronchioles and alveolar ducts are lined by thick hyaline membranes. D: At 72 hours of age, the membranes are being covered by regenerating alveolar lining cells. |
dilated bronchioles and alveolar ducts (Figure 12-32B). Scattered foci of alveolar hemorrhage and edema are present, but most striking is the presence of smooth, homogeneous, pink membranes lining terminal and respiratory bronchioles and alveolar ducts, particularly at points of division or branching (Figure 12-32C). These hyaline membranes are composed of necrotic alveolar lining cells, plasma transudate, inhaled amniotic fluid including squames and fibrin. Hemorrhage is often present. Hyaline membranes may be seen in infants who die as early as 3 to 4 hours after birth and are uniformly present as well-formed structures by 12 to 24 hours in infants with RDS. In the absence of severe disease requiring high oxygen tensions and ventilatory pressures, at 36 to 48 hours, the membranes begin to organize and separate from the underlying wall to be replaced by alveolar lining cells or bronchiolar cuboidal or columnar epithelium (Figure 12-32D). Bacteria may alter the appearance of the membranes by producing fragmented, faintly basophilic structures, with organisms often readily demonstrable by Gram stain on or within the membranes. Conditions associated with hyperbilirubinemia (e.g., kernicterus, intraventricular hemorrhage, intrahepatic bile stasis, disseminated intravascular coagulation) may produce, in infants surviving 3 or more days, yellow hyaline membranes as a result of the presence of unconjugated bilirubin.
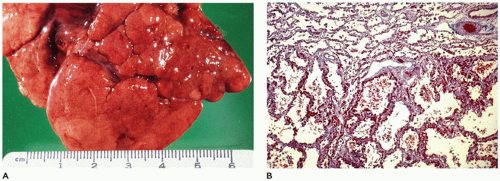 FIGURE 12-33 • Long-standing healed bronchopulmonary dysplasia. A: Irregular clefting and fissuring of pulmonary lobes probably represent the loss of acini during the acute phases of BPD. B: The acinus at top represents the one “protected” by occluded bronchioles from the damage of barotrauma and high oxygen pressures. At the bottom, this acinus displays the diffuse alveolar septal fibrosis caused by previous exposure to barotrauma and high oxygen pressures (Masson trichrome, ×25). |
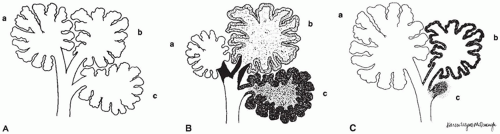 FIGURE 12-34 • Bronchopulmonary dysplasia (BPD) before the advent of surfactant replacement therapy. A: Schematic representation of three uniformly distended acini (a to c) with associated bronchiole, alveolar ducts, and alveoli. B: In the early stages of BPD, hyaline membranes or necrotic debris may totally occlude a bronchiole (a) protecting the distal acinus. Bronchioles that remain partially or completely open (b, c) allow the distal acinus to be exposed to varying degrees of injury from barotrauma and high oxygen tension. C: In the healed stages of BPD with resolution of the bronchiolar obstruction in (A), the “protected” distal acinus expands and continues to develop new alveoli. Depending on the degree of injury, acini may atrophy and disappear (c), producing pleural fissures (see Figure 12-33A), or display varying degrees of alveolar septal fibrosis (b) and be inhibited from further alveolar development. (From Stocker JT. Pathologic features of long-standing “healed” bronchopulmonary dysplasia: a study of 28 3- to 40-month-old infants. Hum Pathol 1986;17:943, with permission.) |
it is possible that low levels of oxygen (25% to 35%), while not producing significant alteration in the epithelial lining of the lung or not causing damage sufficient to cause septal fibrosis, may, in very immature infants, inhibit growth of the lung, that is, the development of new alveolar ducts and alveoli. Although the lung appears to “mature” and alveolar septa appear to thin and expand to resemble the septa of term infants, there is no accompanying significant increase in the surface area of the lung through an increase in number of alveoli. Thus, although recent advances in mechanical ventilation have limited the amount of injury to the bronchiole (i.e., no necrotizing bronchiolitis), the continued patency of all bronchioles throughout the course of therapy allows equal injury or inhibition of growth to all acini from even low levels of oxygen therapy.
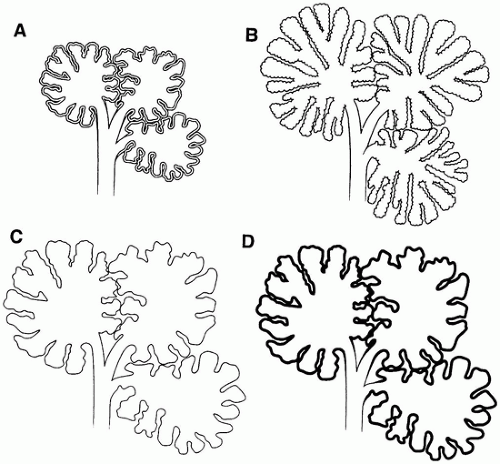 FIGURE 12-35 • Chronic lung disease of the premature (CLDP) since the advent of surfactant replacement therapy. A: Schematic representation of three normally expanded and aerated pulmonary acini in an immature infant. Note the appropriately thick septa of the developing lung. B: With normal lung growth and development, the acini not only increase in size [relative to (A)] but also in complexity with the appearance of “new” alveolar saccules and alveoli. C and D: In infants receiving surfactant replacement therapy who develop moderate-to-severe CLDP, the acini increase in size [relative to (A)] but show little, if any, increase in the number of alveolar saccules or alveoli. The alveolar septa in (C) appear normal in thickness compared with the less-injured or uninjured lung (B), or they may display a uniform mild alveolar septal fibrosis as in (D). |
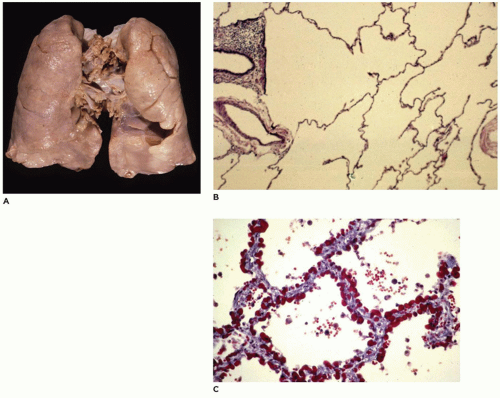 FIGURE 12-36 • Chronic lung disease of the premature. A: The lung appears largely unremarkable with an evenly aerated parenchyma. B: In this section of lung from a 4-month-old infant born at 26 weeks gestation who developed moderate respiratory distress and clinical BPD, the acini are simplified with dilated alveolar ducts and saccules and with very few alveoli arising from them. C: In this section of lung from a 2-month-old infant born at 28 weeks gestation who developed severe prolonged respiratory distress and clinical BPD, the alveolar septa of all acini show mild though uniform interstitial fibrous thickening.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|