The quantum mechanical description of nature
The purpose of this chapter is to establish the mathematical foundations of quantum theory. In the previous chapter we became acquainted with ‘old’ quantum theory. This theory resembles the structure of quantum theory but it does so with ad hoc assumptions. Now, we will attempt to introduce a set of postulates – fundamental statements of fact that cannot be derived from more basic tenets – from which all other aspects of quantum theory are derived. In doing so we hope to establish reasons for the assumptions made in ‘old’ quantum theory. These postulates should also allow us to derive new results and make predictions about previously unknown phenomena.
This chapter may appear overly mathematical and formal. And it is. That is the nature of the foundations of quantum mechanics. This is essential hard work. However, the purpose of this chapter is not just to make you familiar with the mathematics of quantum mechanics. It is also meant to make you familiar with the vocabulary and concepts of quantum mechanics. Read it out loud to yourself – preferably alone, as muttering about quantum mechanics in public spaces is not necessarily the best way to win friends and influence people!
20.1 What determines if a quantum description is necessary?
We have already encountered one principle to guide us in determining the necessity for a quantum description:
We can expect to observe quantum mechanical effects and the insufficiency of a classical mechanical description when at least one dimension of the system under consideration approaches the magnitude of its de Broglie wavelength.
A second condition for the necessity for the use of quantum mechanics rather than classical mechanics can be stated as so:
A quantum mechanical description should be used when the thermal energy kBT is small compared to the energy level spacing ϵi − ϵj.
Classical mechanics can be derived from quantum mechanics in the limit in which allowed energy values are continuous rather than discrete. As shown by Ehrenfest, this occurs mathematically in the limit of ℏ approaching zero. In a practical sense, this occurs when the thermal energy kBT is large compared to the energy level spacing ϵi − ϵj between successive levels i and j. You will recall from our discussion of statistical mechanics in Chapter 2 that a thermal distribution is characterized by a Boltzmann distribution,

Classically, this is a continuous distribution, but quantum mechanically this is a discrete distribution. For H2 gas filling a macroscopic container at room temperature, the translational degree of freedom acts decidedly classically because the spacing between translational levels is much smaller than the thermal energy. The rotational and vibrational degrees of freedom, on the other hand, display characteristics of their quantum state distributions that are readily detected by spectroscopy, as we will explore in more detail in the chapters to follow.
The above statements are, of course, expressions of Bohr’s correspondence principle:
In the limit of high quantum numbers, systems behave classically. Equivalently, in the limits of large energies, large masses and large orbits, quantum calculations deliver the same results as classical calculations.
Thus, it is in the opposite limits (low temperature, small mass and small length scale) in which quantum calculations are unavoidable.
20.2 The postulates of quantum mechanics
The laws of thermodynamics cannot be derived from more fundamental principles. They simply are the basis of thermodynamics from which the remainder of the theoretical framework of thermodynamics is derived. Similarly, the postulates of quantum mechanics cannot be derived from more fundamental principles. They form the basis of the theory from which its framework is derived. As with the laws of thermodynamics, I present the postulates first and then we will delve into their meaning and implications in subsequent chapters. The content of the postulates is more or less resolved. However, unlike the laws of thermodynamics, the numbering (and even the number) of the postulates of quantum mechanics is not set by convention. Reading the postulates, you will see that we need to define several terms including eigenfunction, eigenvalue, Hermitian and operator. This we will do in the remainder of the chapter.
Postulate 1: Completeness and Repeatability
Every physical system of N particles is completely described by a mathematical function of the coordinates τ1, τ2,…, τN of all the particles in the system and of time t. This finite, continuous and single-valued function is called the system’s wavefunction, Ψ(τ1, τ2…, τN, t). The state described by this wavefunction is a predictive tool. An immediately repeated measurement yields the same result as long as the state described by this wavefunction is an eigenfunction.
Postulate II: Born’s Rule
The wavefunction is a probability amplitude that potentially contains a phase factor (an imaginary part). Therefore, the probability distribution described by the wavefunction is obtained from its absolute square and Ψ(τ, t)*Ψ(τ, t)dτ is proportional to the probability that the system is found in the range dτ about τ at time t.
Postulate III: Collapse Axiom A
Each physically observable quantity is represented mathematically by a unique operator that acts on the wavefunction in a prescribed manner. Operators that correspond to physical properties must be linear and Hermitian and, therefore, the eigenstates of the operator (and wavefunctions that describe the system) must be orthogonal.
Postulate IV: Collapse Axiom B
A wavefunction ψ that describes a system forms an eigenvalue equation with the operator
such that
where α is the value of the physical quantity represented by
that would be measured experimentally for the system. Thus, any system whose wavefunction is an eigenfunction of
has a definite value of α.
Postulate V: Unitarity
To every physical system there corresponds a unique operator representing the total energy of the system. This operator, the Hamiltonian
, and the observable physical states of the system are described by those wavefunctions that satisfy the time-dependent Schrödinger equation

Postulate VI: Expectation Values
Consistent with the correspondence principle and the interpretation of the wavefunction as a probability amplitude, the average measurement of a physical observable is represented by the quantum analog of the classical mean of the operator
operating on the wavefunction ψ according to
. ⟨A⟩ is known as the expectation value of the operator
.
Postulate VII: Superposition Principle
Since the eigenfunctions of a given Hamilitonian form a complete set of orthogonal, normalizable functions, they correspond to vectors in a Hilbert space such that, for any two good wavefunctions ψ1 and ψ2, the superposition state represented by ψ = c1ψ1 + c2ψ2 is also a good wavefunction.
Postulate VIII: Composition Postulate
Composite states, such as those formed between the system S and the environment E can be expressed as superpositions of the form
where ψs,k and ψs,l are the basis functions that describe the system and the environment.
In the following chapters we will deal with all of these postulates directly, even briefly in the case of Postulate VIII, which is the basis for entanglement. The phenomenon of entanglement brings to light some rather fundamental paradoxes of quantum mechanics in which quantum behavior simply does not comport with our classical experiences1. Schrödinger identified it as “…the characteristic trait of quantum mechanics, the one that enforces its entire departure from classical lines of thought.”2 These issues are embodied in the so-called Einstein–Podolsky–Rosen paradox3 and tests of Bell’s inequalities4. This postulate has deep implications for the meaning of and our perception of reality. An excellent discussion of quantum paradoxes is given by Griffiths5. While we will not deal with the mathematics of entanglement in detail, we will return to the topic when we discuss photosynthesis as entanglement and long-lived coherent excitation transfer appear to play a role in photosynthesis in the process of transferring the energy of a photon absorbed in a light-harvesting center to the reaction center6.
20.3 Wavefunctions
20.3.1 Mathematical requirements
Postulate I requires the existence of a wavefunction, and physical reality constrains the mathematical nature of these functions. As we shall see below, Postulate V allows us to write the complete wavefunction, which is a function of space and time, as the product of a function of the spatial coordinates alone and a time-dependent function,

(as long as the potential is independent of time). The composite variable τ represents the spatial coordinates of each particle in the system. The physical constraints on the wavefunction lead to the following set of mathematical boundary conditions:
- Ψ must be continuous, finite (contain no singularities) and single-valued.
- Ψ is a function that may contain complex numbers.
- Ψ must be quadratically integrable and normalizable.
- All partial first derivatives of Ψ must be continuous (except for when an infinitely high potential barrier is encountered), contain no singularities, and be single-valued.
We shall see shortly that the time-dependent function is the simple exponential function φ(t) = exp (Et/iℏ), hence, all of the constraints on the spacetime wavefunction Ψ also apply to the spatial wavefunction ψ.
A continuous function is smooth, containing no jumps. A jump corresponds to an infinite first derivative. Since the linear momentum of a particle is proportional to the first derivative of the wavefunction, an infinite first derivative implies an infinite momentum. This is impossible in any realistic physical system.
The value of the wavefunction must be finite even at infinity. In fact, the wavefunction must tend to zero as it approaches infinity, otherwise its integral would not be finite. A single-valued wavefunction can have one, and only one, value at any set of coordinate values. A complex number contains both a real and an imaginary part. Postulate II requires the product of a wavefunction with itself to result in a real number. This can only occur in general for complex numbers if we form the product between a function and its complex conjugate in which i is replaced by –i in all imaginary terms. This product is denoted the absolute square of the wavefunction |Ψ|2 = ψ*ψ. Furthermore, since we need to integrate this product to determine the probability of the existence of the state, the wavefunction must be quadratically integrable to a finite real number. If the wavefunction is quadratically integrable to a finite real number, then we can always scale it such that the wavefunction is normalized, that is, ∫∞− ∞ψ*ψ dτ = 1.
Finally, Ψ will be continuous and smooth except for at a finite number of points that will be clearly identifiable within the model we are using (such as when the potential goes to infinity, as it does at the infinitely high walls of the particle in a box problem below); hence, its partial first derivatives are also continuous except at certain boundaries imposed by the model. A discontinuity in the first derivative implies an infinite second derivative. Since the energy operator is proportional to the second derivative of the wavefunction, an infinite second derivative implies an infinite energy, which is not physically realistic.
20.3.2 Born interpretation of the wavefunction
Schrödinger proposed the wave equation that we examine in detail below without really understanding what the wavefunction was. Initially, it was unclear whether this function was merely a mathematical construct that delivered the correct answers, or whether there was a deeper physical meaning to the wavefunction. It was Born who realized how the wavefunction could be interpreted physically7. His proposal was that ψ is a probability amplitude and its absolute square

is proportional to the probability of finding the particle at position x. |ψ(x)|2 is a probability density, as illustrated in Fig. 20.1.

Figure 20.1 Illustrations of ψ and |ψ|2 for the n = 4 state of a particle in a box. A probability amplitude can be either positive or negative, which is related to the potential for interference effects upon combination with another wavefunction. The probability density is always positive, and its integral is proportional to the probability of finding the particle between the limits of integration. By symmetry, it is easy to see that the integral of the blue region is 0.25 of the normalized area of the complete integral of |ψ|2.
The implications of the Born interpretation are rather unsettling for someone steeped in the seeming determinism of classical mechanics. If a particle is described by ψ(x) and its position by a probability distribution, then the particle must have wave-like character and the concept of a single well-defined trajectory has to be abandoned. This will become all the more disruptive to classical intuition when we encounter further consequences that are known as the Heisenberg uncertainty principle.
Linking the square of the wavefunction to a probability density means that the product of the square of the wavefunction times a volume is equal to the probability of finding the particle in that volume. Thus, the probability of finding a particle in a certain region of space between τ1 and τ2 is

If there is one particle described by the wavefunction, then there is unit probability of finding that particle when considering all of the space in which that particle can be found. In other words, a good wavefunction is normalized to 1 when integrated over all space8. The normalization condition is that we should be able to scale our wavefunctions by a factor N such that when the square of the wavefunction is integrated over all space, the result is unit probability,

The volume element dτ depends on the coordinates and dimensionality. Assuming that we have properly normalized the wavefunction and incorporated the constant N into ψ, in one-dimensional linear coordinates, the normalization condition is
1D, linear coordinates

while in three dimensions it reads
3D, linear coordinates

For central field problems, such as an atom, spherical polar coordinates are more appropriate,
3D, spherical polar

Recall that if a function is variable separable and can be factored such as,

then the integrals can also be factored, for example,

Both 1 and N are dimensionless quantities. Since the normalization integral depends on the dimensionality of the system, the dimensions of ψ must also change with the dimensionality of the system.
20.3.2.1 Example: particle in a one-dimensional box
The wavefunction for a particle confined in a one-dimensional box of length L, as we shall prove in the next chapter, is (in the range 0 ≤ x ≤ L)

Take L = 10.0 nm. Now let’s calculate the probability that a ground-state electron (that is, an electron in the n = 1 state) is (a) between x = 4.95 nm and 5.05 nm, and (b) in the left half of the box. (c) What is the probability of finding the particle at x = 0 or x = 5 nm?
The n = 1 wavefunction is ψ = (2/L)1/2sin (πx/L). The probability is found by integration of the square of the wavefunction over the appropriate limits.

- In this case a = 4.95 nm and b = 5.05 nm.

Substituting for a and b we find P = 0.010.
- In this case a = 0 and b = L/2 = 5. However, since the wavefunction is symmetric about the center of the box and the integral over the whole of the box is 1 (normalized wavefunction), the integral over half of the box is 0.5. Look back at Fig. 20.1. The n = 4 state is shown, which has four extrema and two full cycles of the sine function. The n = 1 state corresponds to just one half-cycle of the sine function.
- The wavefunction is zero at x = 0. The probability is also zero at x = 0. The wavefunction is a maximum at x = 5 nm; nonetheless, the probability of finding a particle at a point – any point – is zero. The integral of any finite function over a single point is zero9. Ponder that for a moment. The particle does not exist at any point in the box. Yet the particle is in the box. Ergo, the particle is not point-like. The probability of finding the particle in any region of space is nonvanishing. In the vicinity of a node, this probability is exceedingly small. The smaller we make the region, the smaller the probability, but it only goes uniquely to zero at a point. Thus, there is no difficulty for this non-point-like particle to move from one side of the box to the other, even though the wavefunction contains nodes in it.
20.4 The Schrödinger equation
The wave-like nature of small particles means that they do not follow the well-defined trajectories inherent to classical mechanics. Instead, quantum mechanics uses wavefunctions to describe particles and their motions. A wavefunction describes a probability amplitude and the square of a wavefunction tells us something about the probability of a particle being at a certain point in space and time. We must abandon a description in terms of particle trajectories, or – as conceived of in the path integral formulation of quantum mechanics – all trajectories are potentially possible for the particle, but they are not equally probable. We need to learn how to calculate the probabilities and to understand the implications of particle dynamics being described by a probability amplitude that contains both magnitude and phase information.
The Schrödinger equation was proposed by Schrödinger10 in 1926. His argument for what was at the time a very preposterous idea – that we really do have to take this wave-particle duality of de Broglie seriously when discussing the dynamics of subatomic particles – was to state that geometrical optics (treating light as rays) and physical optics (treating light as waves) emerge from one another in different limits. Furthermore, William Hamilton used as an analogy the theory of the propagation of light in a nonhomogeneous medium to derive theories in pure mechanics, most notably the Hamilton function of action and how it is used to describe the evolution of a mechanical system. It was left to Schrödinger to reverse engineer from Hamilton’s work the implications for atomic-scale systems. In doing so he would derive the Planck relation of energy E = ℏω, Bohr’s energy level structure of the H atom En = −2π2me4/h2n2, and both the intensities and polarizations of spectral lines observed in the Stark effect as natural consequences of these dynamics. Something this good just could not be wrong, so he forged ahead without a clear conception about the nature of the wavefunction.
For a particle of mass m moving in one dimension x with a possibly time-dependent potential described by V(x,t), the time-dependent Schrödinger equation has the form

The time-dependent wavefunction Ψ(x,t) is the solution to this equation and ℏ = h/2π is the reduced Planck constant. In many of our applications, the potential is independent of time. This allows us to factor the wavefunction into a spatial component and a time-dependent component,

The function ψ(x) is the spatial part of the wavefunction. These functions are also called the stationary states of the system.
Upon substitution of the factored wavefunction back into Eq. (20.11), the time-dependent Schrödinger equation becomes

Each side of Eq. (20.13) is a function of one variable only: x for the LHS and t for the RHS. They can only be equal to each other if they are both equal to the same constant. We call this constant E, and we will discover that it is equal to the total energy of the state described by the wavefunction. Setting the RHS equal to E, we obtain,

The solution to this differential equation is the exponential function,

which describes a sinusoidally oscillating wave with angular frequency ω = E/ℏ. For a potential that does not vary in time, the total energy E = ℏω is constant and the time-dependent wavefunction has the form

From the Born interpretation, we learn that the wavefunction is a probability amplitude, the absolute square of which gives the probability of finding the particle. However, to take the absolute square, we must take into consideration that the wavefunction is a complex function; thus, the square of the function is taken by multiplying the complex conjugate of the wavefunction by the wavefunction,

The complex conjugate is taken by switching the sign of each term involving an imaginary number i → −i; hence,

This explains why the ψ(x) are called the stationary states – the probability distribution described by the square of the wavefunction is not time-dependent.
The LHS of Eq. (20.13) is also equal to the total energy E. This allows us to write the time-independent Schrödinger equation,

We introduce the Hamiltonian operator 

which allows us to write the time-independent Schrödinger equation in the compact form

Most of the questions we will try to answer will have their solution in the time-independent Schrödinger equation, which is why the short form in Eq. (20.21) has become so familiar. Essentially all of chemistry and physics is contained within the Schrödinger equation. This statement comes with the two very significant ‘howevers.’ The first is that we need to write down a complete Hamiltonian to describe all the interactions in our system. The kinetic energy term is easy, but the potential energy term requires detailed knowledge and a mathematical expression for every kind of interaction (electrostatic, magnetic, gravitational, chemical, van der Waals, etc.) experienced in the system. Once we can formulate all of the interactions, ‘all’ we have to do is find the wavefunctions. However – and this one is even tougher – the Schrödinger equation cannot be solved exactly for any atom or molecule containing more than one electron. Obviously, a major component of quantum mechanics is the pursuit of ever better approximations to the exact solution of the Schrödinger equation.
20.4.1 Directed practice
The time-dependent Schrödinger equation is sometimes written

Show that this is equivalent to Eq. (20.16).
20.5 Operators and eigenvalues
Postulates III and IV intentionally mention operators and eigenvalues. To mathematicians, these are terms that are loaded with meaning invoking a Pavlovian response. They are built into the language of the postulates because they bring with them immediate clues as to how to formulate and solve the Schrödinger equation.
An operator is something that carries out mathematical manipulations on the function ψ. For example, to find the momentum, apply the momentum operator  to the wavefunction that describes the system. The hat (or circumflex) on top of
to the wavefunction that describes the system. The hat (or circumflex) on top of  (read ‘p-hat’) is there to differentiate the linear momentum operator
(read ‘p-hat’) is there to differentiate the linear momentum operator  from the magnitude of the linear momentum p, just as using bold-italics differentiates the linear momentum vector p from its magnitude. To find the energy, we apply the energy operator, given the special name of the Hamiltonian
from the magnitude of the linear momentum p, just as using bold-italics differentiates the linear momentum vector p from its magnitude. To find the energy, we apply the energy operator, given the special name of the Hamiltonian  because of its singular importance.
because of its singular importance.  is a second-order differential operator. It contains terms like d2/dx2 but not higher derivatives. The form of
is a second-order differential operator. It contains terms like d2/dx2 but not higher derivatives. The form of  depends on the system. We have to consider all factors that contribute to energy – both potential and kinetic – to construct the Hamiltonian.
depends on the system. We have to consider all factors that contribute to energy – both potential and kinetic – to construct the Hamiltonian.
Whenever an operator is involved in an equation, the order of operation is important. For instance, in Eq. (20.21) we cannot first factor out ψ and then operate. We must first operate, and then solve the resulting equation. The order of operation is also indicated in the operator. The operator x(d/dx) means first take the derivative with respect to x, then multiply by x. The operator (d/dx)x means first multiply by x and then take the derivative with respect to x. Note that the order of operation is not important for multiplication or division by constants. In addition, operators operate on functions, not on other operators. Hence,  means operate first with
means operate first with 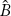 on ψ, then with
on ψ, then with  on the function that results from
on the function that results from 
The order of operation is important because operators may or may not commute. Operators are said to commute if, for all ψ,

Operators do not commute if, for all ψ,

The difference has profound implications. Because of this, we define the commutator of  and
and  , which is itself an operator, as
, which is itself an operator, as

When applied to a function, the commutator is evaluated by operating on ψ with  then subtracting from this the result of operating on ψ with
then subtracting from this the result of operating on ψ with  ,
,

Operators act on functions to produce new functions. Postulate III stipulates that the operators are linear, which means that they obey the following rules

and

where c is a constant, again reaffirming that the order of operation for multiplication (or division) by a constant is unimportant.
Postulate III also stipulates that the operators are Hermitian. An operator is Hermitian if for two functions ψ1 and ψ2

For operators that correspond to physically observable quantities, the above two integrals are equal. The LHS corresponds to operating first on ψ2 then multiplying by the complex conjugate of ψ1. The RHS corresponds to operating on ψ1, taking the complex conjugate then multiplying by ψ2. It is important that the operators are Hermitian because Hermitian operators have real eigenvalues.
20.5.1 Example
Show that the eigenvalue α of a Hermitian operator  operating on the normalized wavefunction ψ is real.
operating on the normalized wavefunction ψ is real.
According to the given set of assumptions, our operator and wavefunction must satisfy the eigenvalue equation

Now, multiply both sides of this eigenvalue equation from the left with ψ* and integrate over all space,

α is a constant and can be moved outside of the integral,

The wavefunction is normalized, which means that the integral on the right is equal to 1,

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


