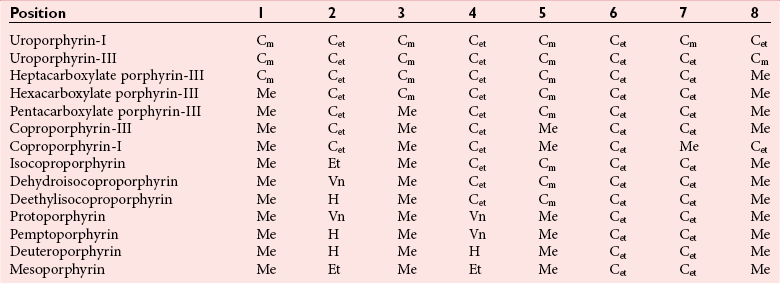
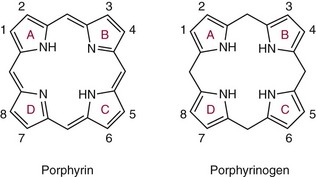
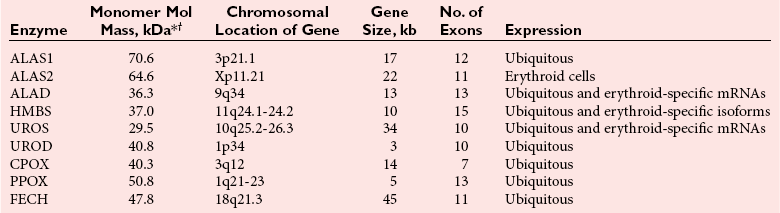
Chapter 33 Michael N. Badminton, M.B., Ch.B, Ph.D., F.R.C.Path., Sharon D. Whatley, Ph.D., Allan C. Deacon, Ph.D., F.R.C.Path. and George H. Elder, M.D., F.R.C.P., F.R.C.Path. The porphyrias are a group of uncommon, inherited disorders of heme biosynthesis.3,102 Each porphyria results from a partial deficiency of one of the enzymes of the pathway converting 5-aminolevulinate (ALA) to heme or, in one rare disorder, an increase in activity of the rate-controlling enzyme of erythroid heme synthesis. Each functional abnormality is associated with a specific pattern of overproduction, accumulation, and excretion of the intermediates of the pathway. These intermediates are excreted in excessive amounts in urine, feces, or both. The clinical consequences depend on the nature of the heme precursors that accumulate. In the acute porphyrias, excess porphyrin precursors [ALA and porphobilinogen (PBG)] are associated with potentially fatal acute neurovisceral attacks that often are provoked by (1) various commonly prescribed drugs, (2) hormonal factors, (3) alcohol, (4) starvation, (5) stress, or (6) infection. In the nonacute porphyrias, and in those acute porphyrias in which both acute neurovisceral attacks and skin lesions occur, accumulation of porphyrins results in photosensitization of sun-exposed skin. Diagnosis depends on laboratory investigation to demonstrate the pattern of heme precursor accumulation specific for each type of porphyria and requires examination of appropriate specimens for the key metabolites using adequately sensitive and specific methods. Technical advances in the field of molecular genetics have made it possible to investigate all porphyrias at the molecular level. Although rarely essential for diagnosis of symptomatic cases, DNA analysis is now the method of choice for the investigation of families with porphyria and continues to provide new information about the pathology of these disorders. Abnormalities of porphyrin excretion and accumulation also occur in a wide variety of other disorders that are collectively far more common than the porphyrias. Recognition of these secondary porphyrin disorders is important to avoid diagnostic errors. The basic porphyrin structure consists of four monopyrrole rings connected by methene bridges to form a tetrapyrrole ring (Figure 33-1).16 Many porphyrin compounds are known, but only a limited number are of clinical interest. The porphyrin compounds of relevance to the porphyrias (Table 33-1) differ in the substituents occupying peripheral positions 1 through 8. Variation in the distribution of the same substituents around the peripheral positions of the tetrapyrrole ring gives rise to porphyrin isomers, which usually are depicted by Roman numerals (e.g., I, II, III). The reduced form of a porphyrin, known as a porphyrinogen (see Figure 33-1), differs by the absence of six hydrogens (four from the methylene bridges and two from ring nitrogens). Porphyrinogens are unstable in vitro and are spontaneously oxidized to the corresponding porphyrins. Under the lower oxygen tension of the cell, porphyrinogens are sufficiently stable to act as intermediates of the heme biosynthetic pathway; aromatization to protoporphyrin at the penultimate step requires an enzyme. Figure 33-1 Porphyrin and porphyrinogen structures: numbers 1 to 8 represent various substituents, the nature and order of which determine the type of porphyrin or porphyrinogen (see Table 33-1). Numbering system and ring designations are based on the Fischer system. A revised system formulated by the International Union of Pure and Applied Chemistry–International Union of Biochemistry (IUPAC-IUB) Joint Commission on Biochemical Nomenclature is appropriate for more complex needs. Because of increasing complexity, this Roman numeral designation is no longer recommended above IV.86 The complex tetrapyrrole ring structure of heme is built up in a stepwise fashion from the very simple precursors succinyl–coenzyme A (CoA) and glycine (Figure 33-2).21 The pathway is present in all nucleated cells. From measurements of total bilirubin production,8 it has been estimated that daily synthesis of heme in humans is 5 to 8 mmol/kg body weight. Of this, 70 to 80% occurs in the bone marrow and is used for hemoglobin synthesis. Approximately 15% is synthesized in other tissues, mainly the liver and is used to produce cytochrome P450, mitochondrial cytochromes, and other hemoproteins. The pathway is compartmentalized, with some steps occurring in the mitochondrion and others in the cytoplasm. Several carriers that transfer intermediates across the mitochondrial membrane have now been identified.50,112a Although all are potential sites for pathogenic mutations, only one such mutation has been identified: a mutation in the erythroid-specific mitochondrial glycine transporter SLC25A38 that causes nonsyndromic autosomal recessive sideroblastic anemia.50 Figure 33-2 Biosynthetic pathway of porphyrins and heme. Cet, The genes for all enzymes of human heme biosynthesis have been characterized (Table 33-2). The structures of human hydroxymethylbilane synthase (HMBS), uroporphyrinogen-III synthase (UROS), uroporphyrinogen decarboxylase (UROD), coproporphyrinogen oxidase (CPOX), ferrochelatase (FECH), bacterial 5-aminolevulinate synthase (ALAS), 5-aminolevulinic acid dehydratase (ALAD), and protoporphyrinogen oxidase (PPOX) have been determined by x-ray crystallography.5,19,35,44,73,80,131,135 TABLE 33-2 Human Enzymes and Genes of Heme Biosynthesis *ALAD is a homo-octamer, and HMBS and UROS are monomers; all other enzymes are homodimers. †Molecular masses for ALAS1, ALAS2, CPOX, and FECH include presequences that are cleaved during mitochondrial import. HMBS (also known as PBG deaminase) is a cytoplasmic enzyme that catalyzes the formation of one molecule of the linear tetrapyrrole 1-hydroxymethylbilane (HMB; also known as preuroporphyrinogen) from four molecules of PBG with the release of four molecules of ammonia.124 The enzyme has two molecules of its own substrate: PBG, attached covalently to the apoenzyme as a prosthetic group.115 The enzyme is susceptible to allosteric inhibition by intermediates farther down the heme biosynthetic pathway, notably coproporphyrinogen-III and protoporphyrinogen-IX.85 UROS is a cytoplasmic enzyme that rearranges and cyclizes HMB to form uroporphyrinogen-III.124 Each pyrrole ring of HMB contains a methylcarboxylate and an ethylcarboxylate substituent, which are in the same orientation. By the rotation of zero, one, or two alternate or two adjacent pyrrole rings, it is possible to arrive at four different isomers. Apart from closing the ring structure, the enzyme rotates the D-ring via a spirane intermediate,7 producing the type III isomer—an essential reaction because only this isomer contributes to heme biosynthesis. HMB is unstable, and in those porphyrias in which excess HMB accumulates, cyclization occurs nonenzymatically with the formation of the type I isomer. Normally, only minimum amounts of uroporphyrinogen-I are formed. This is the last cytoplasmic enzyme in the pathway, and it catalyzes the decarboxylation of all four carboxymethyl groups to form the tetracarboxylic coproporphyrinogen. The enzyme will use I and III isomers of uroporphyrinogen as substrate. Decarboxylation commences on ring D and proceeds stepwise through rings A, B, and C with formation of heptacarboxylate, hexacarboxylate, and pentacarboxylate intermediates at a single active site.95 Decreased UROD activity causes accumulation of these intermediates in addition to its substrate, uroporphyrinogen. At high substrate concentrations, decarboxylation occurs by a random mechanism.78 CPOX, which is located in the intermembrane space of mitochondria, catalyzes the sequential oxidative decarboxylation of the 2- and 4-carboxyethyl groups to vinyl groups to produce the more lipophilic protoporphyrinogen-IX, with formation of a tricarboxylic intermediate, harderoporphyrinogen.71 Oxygen is required as the oxidant. The enzyme requires sulfhydryl groups for activity, making it a target for inhibition by metals.133 The enzyme is specific for the type III isomer, so that metabolism of the I-series of porphyrins does not occur beyond coproporphyrinogen-I. The product of the enzyme differs from the substrate in that replacement of two of the carboxyethyl groups by vinyl groups introduces a third substituent into the molecule. Therefore the number of possible isomeric forms is increased, and conventionally the numbering system changes, so that the III isomer becomes the IX isomer. In UROD-deficient states, one of the ethylcarboxylate groups of the accumulated pentacarboxylate porphyrinogen is decarboxylated by CPOX to form the isocoproporphyrin series of porphyrins. PPOX, a flavoprotein located in the inner mitochondrial membrane, catalyzes the removal of six hydrogens (four from methylene bridges and two from ring nitrogens) to form protoporphyrin-IX. This involves a three-step, six-electron flavin adenine dinucleotide (FAD)-dependent oxidation that consumes molecular oxygen.21 Nonenzymatic oxidation also occurs in vitro. However, under the low oxygen tension in the cell, PPOX is essential for oxidation to occur. The protoporphyrin produced is the only porphyrin that functions in the heme pathway. Other porphyrins are produced by nonenzymatic oxidation and represent porphyrinogens that have irreversibly escaped from the pathway. FECH (also known as heme synthase) is an iron-sulfur protein located in the inner mitochondrial membrane.135 This enzyme inserts ferrous iron into protoporphyrin to form heme. During this process, two hydrogens are displaced from the ring nitrogens. Other metals in the divalent state also act as substrates, yielding the corresponding chelate (e.g., incorporation of Zn2+ into protoporphyrin to yield zinc protoporphyrin). In iron-deficient states, Zn2+ successfully competes with Fe2+ in developing red cells, so that the concentration of zinc protoporphyrin in erythrocytes increases. Some other dicarboxylic porphyrins also serve as substrates (e.g., mesoporphyrin). Integration of the final stages of erythroid heme biosynthesis may be facilitated by interaction between FECH and proteins involved in iron import.18a Typically, only minute quantities of heme precursors accumulate in the body. The route of excretion largely depends on solubility. The porphyrin precursors ALA and PBG are water soluble and are excreted almost exclusively in urine. Uroporphyrinogen, with eight carboxylate groups, is readily water soluble and is also excreted via the kidney. The last intermediate of the pathway, protoporphyrin (and also protoporphyrinogen), which has only two carboxylate groups, is insoluble in water and is excreted in the feces via the biliary tract. The other porphyrins are of intermediate solubility and appear in both urine and feces. Coproporphyrinogen-I is taken up and excreted by the liver in preference to the III isomer, so that coproporphyrinogen-I predominates in feces and coproporphyrinogen-III in urine. All porphyrinogens in the urine or feces are slowly oxidized to the corresponding porphyrins. Reference intervals for porphyrins and their precursors in urine, feces, and blood are given in Table 33-3. TABLE 33-3 Once in the gut, porphyrins are susceptible to modification by gut flora. The two vinyl groups of protoporphyrin are reduced to ethyl groups, hydrated to hydroxyethyl groups, or removed, giving rise to a variety of secondary porphyrins. Gut flora can also metabolize heme (whether dietary heme, as components from cells sloughed off from the lining of the gut, or heme resulting from gastrointestinal bleeding) to produce a variety of dicarboxylic porphyrins.9 In addition, some bacteria are capable of de novo synthesis of porphyrins. Heme supply in all tissues is controlled by the activity of mitochondrial ALAS, the first enzyme of the pathway. Two isoforms of ALAS are known. The ubiquitous isoform, ALAS1, is encoded by a gene on chromosome 3p21 and is expressed in all tissues. Because it has a half-life of only about an hour, changes in its rate of synthesis produce short-term alterations in enzyme concentration and cellular ALAS activity. Synthesis of ALAS1 is under negative feedback control by heme.82 In the liver, but not most other tissues, ALAS1 is induced by a wide variety of drugs and chemicals that induce microsomal cytochrome P450–dependent oxidases (CYPs). This effect is thought to be mediated mainly by direct transcriptional activation by drug-responsive nuclear receptors,118 rather than occurring secondary to depletion of an intracellular regulatory heme pool as a consequence of use of heme for CYP assembly. Induction of ALAS1 is prevented by heme, which acts by destabilizing messenger ribonucleic acid (mRNA) for ALAS1, by blocking mitochondrial import of pre-ALAS1, and possibly by inhibiting transcription.64 In addition, ALAS1 activity is regulated by PGC-1α; an effect that forms a link between the rate of hepatic heme synthesis and nutritional status.51 The erythroid isoform, ALAS2, is encoded by a gene on chromosome Xq21-22 and is expressed only in erythroid cells. Its activity is regulated by two distinct mechanisms.101 Transcription is enhanced during erythroid differentiation by the action of erythroid-specific transcription factors, and mRNA concentrations are regulated by iron. Iron deficiency in erythroid cells promotes specific binding of iron regulatory proteins to an iron-responsive element in the 5′ untranslated region (UTR) of ALAS2 mRNA with consequent inhibition of translation. The porphyrias are a group of metabolic disorders that result from decreased or, in one rare form of erythropoietic protoporphyria, increased activities of the enzymes of heme biosynthesis3,102 (Table 33-4). All are inherited in monogenic patterns, apart from some forms of porphyria cutanea tarda (PCT) and rare erythropoietic porphyrias associated with malignant myeloid disorders. Each type of porphyria is defined by the association of characteristic clinical features with a specific pattern of accumulation of heme precursors that reflects increased formation of the substrate of the enzyme that is partially deficient or becomes secondarily rate-limiting in that type of porphyria (Table 33-5). Defects that cause porphyria have been identified in all enzymes of the pathway except for ALAS1. Mutations that decrease ALAS2 activity cause nonsyndromic X-linked sideroblastic anemia13; those that increase activity cause an X-linked erythropoietic protoporphyria.129 TABLE 33-4 The Main Types of Human Porphyria AD, Autosomal dominant; AR, autosomal recessive; XLD, X-linked dominant. *Estimated prevalence of clinically overt disease in the United Kingdom. †Skin lesions and neurovisceral crises may occur alone or together. TABLE 33-5 aSlight increase only unless uroporphyrin is present.107 bNot always increased during acute attack. cOther methylcarboxylate-substituted porphyrins are increased to a smaller extent; uroporphyrin is a mixture of type I and III isomers; heptacarboxylate porphyrin is mainly type III. dPBG and ALA may be normal when only skin lesions are present. eCoproporphyrin-III/I ratio may be increased, but increase is usually less than in overt HCP.107 fNot increased in about 40% of patients. gProtoporphyrin bound to globin (if hemolysis is seen in the sample) has a peak at 626 to 628 nm. The porphyrias are characterized clinically by two main features: skin lesions on sun-exposed areas and acute neurovisceral attacks, typically comprising (1) abdominal pain, (2) peripheral neuropathy, and (3) mental disturbance. The skin lesions are caused by porphyrin-catalyzed photodamage, of which singlet oxygen is the main mediator.100 Acute attacks are associated with increased formation of ALA from induced activity of hepatic ALAS1 and partial hepatic heme deficiency, often in response to induction of hepatic CYPs by drugs and other factors. The relationship of these biochemical changes to the neuronal dysfunction that underlines all clinical features of the acute attack is uncertain.76,87 The observation that correction of the metabolic defect in the liver by transplantation is curative113 and that domino transfer of the affected organ to an unaffected recipient causes acute attacks indistinguishable from those suffered by the donor,113a suggests that their primary cause is release of a neurotoxin(s), probably ALA,36 formed in the liver. In Table 33-4, the porphyrias are classified as acute, in which acute neurovisceral attacks occur, or nonacute. Porphyrias also are classified as hepatic or erythropoietic, according to the main site of overproduction of heme precursors. The main hepatic porphyrias are (1) acute intermittent porphyria (AIP), (2) hereditary coproporphyria (HCP), (3) variegate porphyria (VP), and (4) PCT. Erythropoietic porphyrias include congenital erythropoietic porphyria (CEP) and erythropoietic protoporphyria (EPP). Porphyrias also may be classified as cutaneous or acute porphyrias; however, it should be noted that even with these classifications, some porphyrias are difficult to place. The inherited defect in each of the autosomal dominant acute porphyrias (see Table 33-4) is a mutation leading to complete or near complete inactivation of one of the pairs of allelic genes that encode the enzyme whose partial deficiency causes the disorder. Enzyme activities are therefore half of normal in all tissues in which they are expressed, reflecting the activity of the normal gene trans to the mutant allele. Heme supply is maintained at normal or near normal concentrations by upregulation of ALAS1, with a consequent increase in the substrate concentration for the defective enzyme. These compensatory changes vary between tissues; they are most prominent in the liver and are undetectable in most other organs and between individuals. Thus in all autosomal dominant acute porphyrias, some individuals show no evidence of overproduction of heme precursors, and others have biochemically manifest disease with or without clinical symptoms. Low clinical penetrance (the frequency of expression of an allele when it is present in the genotype) is a prominent feature of all the autosomal dominant acute porphyrias.3,102 Family studies indicate that many affected individuals are asymptomatic throughout life. Surveys of blood donors suggest that the AIP gene may be present in as many as 1 in 1675 of the population.90 For all three disorders, the gene frequency is sufficiently high for rare “homozygous” variants of AIP, HCP, or VP to occur in individuals who are homozygotes or compound heterozygotes for disease-specific mutations,32,71 and for the same person to have two separate types of porphyria. About 25% of patients with overt acute porphyria have no family history of the disease. Such sporadic presentation is a reflection of the high prevalence and low penetrance of mutations in the population; acute porphyria caused by de novo mutation is uncommon. All the autosomal dominant acute porphyrias show extensive allelic heterogeneity. More than 340 disease-specific mutations have been identified in the HMBS gene in AIP, about 50 in the CPOX gene in HCP, and more than 150 in the PPOX gene in VP (Human Gene Mutation Database: www.hgmd.org). About 3% of families with AIP have HMBS mutations that only impair expression of the ubiquitous isoform and therefore do not decrease activity in erythroid cells. All other mutations in the autosomal dominant acute porphyrias affect all tissues. Most are restricted to one or a few families, but founder mutations are present in some populations and explain the high frequency of VP in South Africans of Dutch descent and of AIP in Sweden.56,72 Unlike the other acute porphyrias, ADP is an autosomal recessive disorder. Patients are compound heterozygotes or homozygotes for a range of mutations in the ALAD gene.29 The prevalence of heterozygous carriers may be as high as 2% in some populations.79 The life-threatening, acute neurovisceral attacks that occur in AIP, VP, and HCP are clinically identical.3,102 Acute attacks are more common in women, usually occurring first between the ages of 15 and 40, and are very rare before puberty. The main clinical features are summarized in Table 33-6. The clinical features of ADP, which has been reported in only six patients, are similar but may start in childhood.79,102 TABLE 33-6 Clinical Features of an Acute Neurovisceral Attack of Porphyria Data from Elder GH, Hift RJ. Treatment of acute porphyria. Hosp Med 2001;62:422-8. Acute attacks almost always start with abdominal pain that rapidly becomes very severe but is not accompanied by other signs of an acute surgical condition.55,97 Pain may also be present in the back and thighs and may occasionally be most severe in these regions. Signs of autonomic neuropathy, such as vomiting, constipation, tachycardia, and hypertension, are frequent. When convulsions occur, they may be caused by hyponatremia. Pain may dissipate within a few days, but in severe cases a predominant motor neuropathy develops that may progress to flaccid quadriparesis.97 Persistent pain and vomiting may lead to weight loss and malnutrition. The acute phase may be accompanied by mental confusion with abrupt changes in mood, hallucinations, and other psychotic features. However, these mental disturbances disappear with remission. Persistent psychiatric illness is not a feature of the acute porphyrias, although mild anxiety or depression may be present in some patients.88 Abdominal pain usually resolves within 2 weeks, but recovery from neuropathy may take many months and is not always complete. Most patients have one or a few attacks followed by complete recovery and prolonged remission. About 5% have repeated acute attacks, which in women may be premenstrual. Precipitating factors have been identified in about two thirds of patients who present with acute attacks. The most important are (1) drugs; (2) alcohol, especially binge drinking; (3) the menstrual cycle; (4) calorie restriction; (5) infection; and (6) stress. Acute attacks may complicate a small proportion of pregnancies in affected patients. Drugs are frequent precipitants of acute attacks in VP, and hormonal factors appear more important in AIP.55 Drugs known to provoke acute attacks include barbiturates, sulfonamides, progestogens, and some anticonvulsants, but many others have been implicated in the precipitation of acute attacks (http://www.drugs-porphyria.org). Skin lesions similar to those of PCT and other bullous porphyrias are present in about 80% of patients with clinically manifest VP (see Table 33-4). About 60% of patients with this condition present with skin lesions alone. The skin is less commonly affected in HCP; skin lesions without an acute attack are uncommon and usually are provoked by intercurrent cholestasis. Long-term complications of acute porphyria include chronic renal failure, hypertension, and primary hepatocellular carcinoma.3,102 As soon as an attack of acute porphyria is suspected as the cause of illness, drugs and other potential provoking agents should be withdrawn and supportive treatment started using drugs that are known to be safe.34,55,102 Opiates are usually required to control pain. Addition of chlorpromazine or promazine may help to reduce the requirement for analgesics. Patients are prone to severe hyponatremia, and careful administration of any intravenous fluids, with avoidance of hypotonic solutions, is essential. If hyponatremia develops, it should be corrected slowly because patients with acute porphyria are particularly susceptible to cerebral edema and osmotic demyelination. Adequate caloric intake must be maintained, preferably by giving carbohydrate-rich supplements orally or if necessary via a nasogastric tube. When vomiting prevents enteral administration, 100 g of dextrose per day given intravenously as a 5% solution in normal saline should suffice. Unless the attack is mild and is clearly resolving, specific treatment with intravenous heme should be started as soon as the diagnosis has been established.34,55,102 This treatment increases the concentration of heme in the liver, thus decreasing the activity of ALAS1 and the formation of ALA and PBG. The effect of treatment may be monitored by measuring these metabolites, but this is not essential because clinical improvement is the required end point. Heme administration will not reverse an established neuropathy. If heme preparations are not available, hepatic ALA synthase activity can be decreased by carbohydrate loading,14 but this treatment is less effective than intravenous heme and is more difficult to administer. Repeated attacks are difficult to control. Cyclic premenstrual attacks in women may be prevented by suppression of ovulation with gonadorelin analogs, but many patients require repeated courses of intravenous heme.3,34,102 Orthotopic liver transplantation leads to immediate and prolonged remission with restoration of PBG excretion to normal.113 Diagnosis of autosomal dominant acute porphyria should be followed by investigation of the patient’s family to identify affected, often asymptomatic, relatives, so that they can be advised to avoid drugs and other factors known to provoke potentially fatal acute attacks (http://www.drugs-porphyria.org). Presymptomatic diagnosis also has the benefit that specific treatment can be started promptly if an attack does develop without delay while a diagnosis is sought. Although attacks are very rare before puberty, children should be tested at as young an age as is practicable to ensure that their status is known by the time they reach puberty and to enable the very low risk for affected children to be further reduced. Counseling to reduce the risk of an acute attack should include comprehensive information about the disease, including specific advice to guide selection of safe drugs, and provision of jewelry or some other means to identify the individual as having an acute porphyria. Where available, patients should be made aware of the relevant national patient support group. The nonacute porphyrias include PCT, CEP, and EPP. PCT is by far the most common porphyria.31 The annual incidence of new cases in the United Kingdom is between 2 and 5 per million of the population. The disease occurs at all ages in both sexes with onset usually during the fifth and sixth decades. Lesions on sun-exposed skin, particularly the backs of the hands, the forearm, and the face, are present in all patients. These lesions are identical to those seen in the other bullous porphyrias (see Table 33-4). Increased mechanical fragility of the skin, with trivial trauma leading to erosions, is present in virtually all patients. Subepidermal bullae, milia, hypertrichosis of the face, and patchy pigmentation are also common. This combination of skin lesions with liver damage is strongly associated with alcohol abuse, estrogens, infection with hepatotropic viruses, particularly hepatitis C (HCV), and mutations in the hemochromatosis (HFE) gene.15,30,31 PCT may complicate HIV infection.30 Hepatic iron overload and at least one of the other associated factors are present in almost all patients. Between 8% and 79% of patients have antibodies to HCV, with prevalence highest in the United States and southern Europe and lowest in Western Europe. About 20% of patients of Northern European descent are homozygous for the C282Y mutation in the HFE gene, but in spite of having the genotype of genetic hemochromatosis, very few patients have clinical signs or symptoms of iron overload. However, increased serum ferritin concentrations and other biochemical indicators of iron overload are common in PCT irrespective of the HFE genotype, suggesting that the origin of hepatic iron overload is multifactorial. PCT may occur in association with other disorders, notably chronic renal failure, systemic lupus erythematosus, and hematologic malignancies. In addition, rare cases of a PCT-like syndrome resulting from production of porphyrins by primary hepatic tumors have been described.91 PCT results from a decrease in activity of UROD in the liver, which leads to overproduction of uroporphyrin and other carboxymethyl-substituted porphyrinogens. These auto-oxidize to porphyrins, which accumulate in the liver and skin, where they act as photosensitizers and are excreted in urine and bile. Two main types of PCT can be identified by measurement of UROD activity in liver and extrahepatic tissue, and by analysis of the UROD gene. About 80% of patients have the sporadic (type I) form of PCT, in which the enzyme defect is restricted to the liver and the UROD gene appears to be normal. Typically, no family history of PCT is reported, but rare cases are clustered in families (type III PCT). The rest have familial (type II) PCT. In this form, mutation of one UROD gene leads to half-normal UROD activity in all tissues, which is inherited in an autosomal dominant manner. As with the other autosomal dominant porphyrias, clinical penetration of familial PCT is low with considerable allelic heterogeneity; each of the more than 85 mutations is described as present in only one or a few families, except in Norway, where the high prevalence of PCT has been attributed to a founder effect.1 A rare variant of familial PCT, hepatoerythropoietic porphyria (HEP), in which UROD mutations, some of which have also been found in familial PCT, are present on both alleles, has been described.31,32 PCT may also be caused by exposure to certain polyhalogenated aromatic hydrocarbons, such as hexachlorobenzene and 2,3,7,8-tetrachlorodibenzo-p-dioxin.31 In families with familial PCT, a decrease of 50% in enzyme activity is not by itself sufficient to cause clinically overt disease. Further inactivation of UROD in the liver seems to be required, and the process responsible for this inactivation appears also to be responsible for inactivation of hepatic UROD in sporadic PCT and in toxic PCT caused by chemicals. The inactivation process decreases catalytic activity without impairing enzyme concentration, is iron dependent, and is reversible. Current evidence from experimental models of PCT suggests that UROD is inactivated by a porphomethene inhibitor that is produced by iron-dependent oxidation of a substrate of the UROD reaction, possibly mediated by hepatic CYPs, particularly CYP1A2.93 In addition to protection of the skin from sunlight, two specific treatments may be used for PCT: depletion of hepatic iron stores by repeated phlebotomy or other means, and low-dose oral chloroquine.110 In patients with chronic renal failure and PCT, hepatic iron stores can be decreased by erythropoietin with or without phlebotomy. CEP is the least common but most severe of the cutaneous porphyrias.38,121 The prevalence is less than one per million in the United Kingdom. This disorder is also known as Günther disease. The clinical features vary in severity from hydrops fetalis with death in utero—through onset in infancy of severe skin lesions with transfusion-dependent hemolytic anemia—to mild skin lesions, resembling PCT, that do not start until adult life. Late-onset cases may also develop in association with hematologic malignancy, particularly myelodysplasia.65
The Porphyrias and Other Disorders of Porphyrin Metabolism
Porphyrin Chemistry
Structure and Nomenclature
Heme Biosynthesis
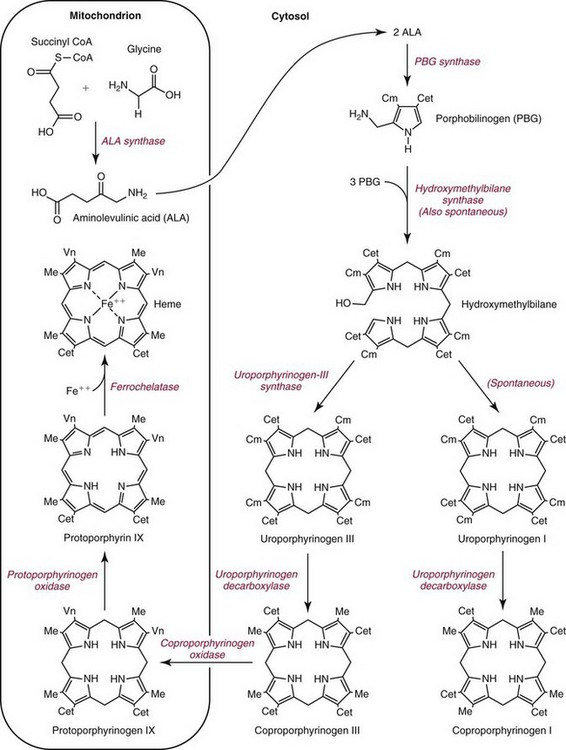
 CH2CH2COOH; Cm,
CH2CH2COOH; Cm,  CH2COOH; Me,
CH2COOH; Me,  CH3; Vn,
CH3; Vn,  CH
CH CH2.
CH2.
Enzymes of Heme Biosynthesis
Hydroxymethylbilane Synthase (EC 2.5.1.61) (HMBS)
Uroporphyrinogen-III Synthase (EC 4.2.1.75) (UROS)
Uroporphyrinogen Decarboxylase (EC 4.1.1.37) (UROD)
Coproporphyrinogen Oxidase (EC 1.3.3.3) (CPOX)
Protoporphyrinogen Oxidase (EC 1.3.3.4) (PPOX)
Ferrochelatase (EC 4.99.1.1) (FECH)
Excretion of Heme Precursors
Specimen
Analyte
Reference Interval
Urine
Porphobilinogen
<10 µmol/L24
<1.5 µmol/mmol creatinine11
Total porphyrin
20-320 nmol/L24
<35 nmol/mmol creatinine11
Uroporphyrin
0.8-3.1 nmol/mmol creatinine9
Heptacarboxylate porphyrin
<0.9 nmol/mmol creatinine9
Coproporphyrin-I
1.2-5.7 nmol/mmol creatinine9
Coproporphyrin-III
4.8-23.8 nmol/mmol creatinine9
% Coproporphyrin-III*
68-869
Feces
Total porphyrin
10-200 nmol/g dry wt77
Coproporphyrin-I
1.1-5.5 nmol/g feces9
Coproporphyrin-III
0.2-2.5 nmol/g feces9
Coproporphyrin-III/I ratio
0.3-1.4130
Total dicarboxylate porphyrin
0.5-12.8 nmol/g feces9
Erythrocytes
Total porphyrin
0.4-1.7 µmol/L erythrocytes
Regulation of Heme Biosynthesis
The Porphyrias
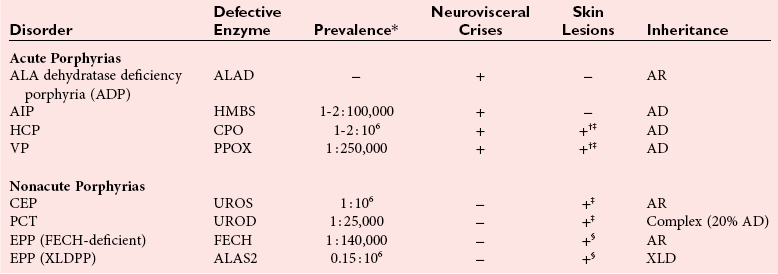
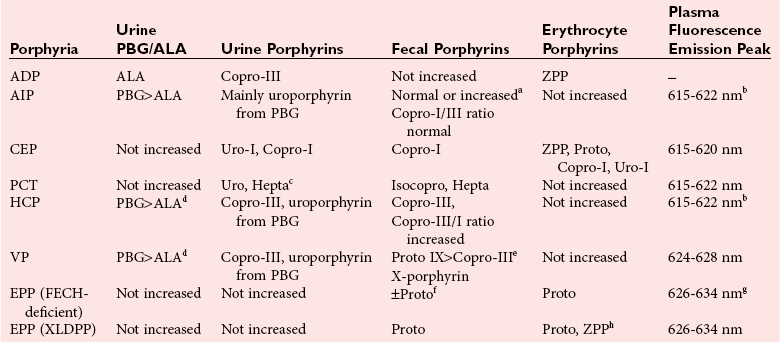
Acute Porphyrias
Biochemistry and Molecular Genetics
Clinical Features
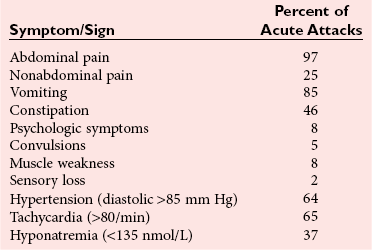
Treatment
Management of Families
Nonacute Porphyrias
Porphyria Cutanea Tarda
Clinical Features
Pathogenesis and Molecular Genetics
Treatment
Congenital Erythropoietic Porphyria
Clinical Features
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The Porphyrias and Other Disorders of Porphyrin Metabolism