8
CHAPTER OUTLINE
Sedative–hypnotic drugs represent a diverse group of chemical agents that suppress central nervous system (CNS) activity (1). They are used in medicine as anxiolytics, hypnotics, anticonvulsants, muscle relaxants, and anesthe-sia induction agents. Their calming effects are on a continuum with sleep-inducing effects, unconsciousness, and, for some agents, coma and death. Substances discussed in this chapter include benzodiazepines, nonbenzodiazepine hypnotics, barbiturates, and miscellaneous, related compounds.
FORMULATIONS AND CHEMICAL STRUCTURE
The basic structure of the benzodiazepines is the 1,4-benzodiazepine nucleus as shown in Figure 8-1 (2). Various substitutions alter the efficacy, potency, and other properties of individual drugs (3). Variations on the benzodiazepine ring structure have produced (a) the triazole group: alprazolam, triazolam, and estazolam; (b) the 2-keto group: diazepam, flurazepam, and clorazepate; (c) the 2-amino group: chlordiazepoxide; (d) the 3-hydroxy group: lorazepam, oxazepam, and temazepam; (e) the trifluoroethyl group: quazepam; (f) the imidazole group: midazolam; and (g) the 7-nitro group: nitrazepam and clonazepam.
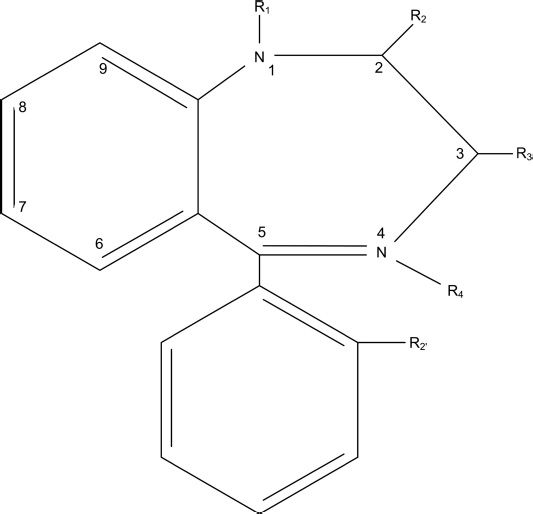
FIGURE 8-1 The 1,4-benzodiazepine nucleus.
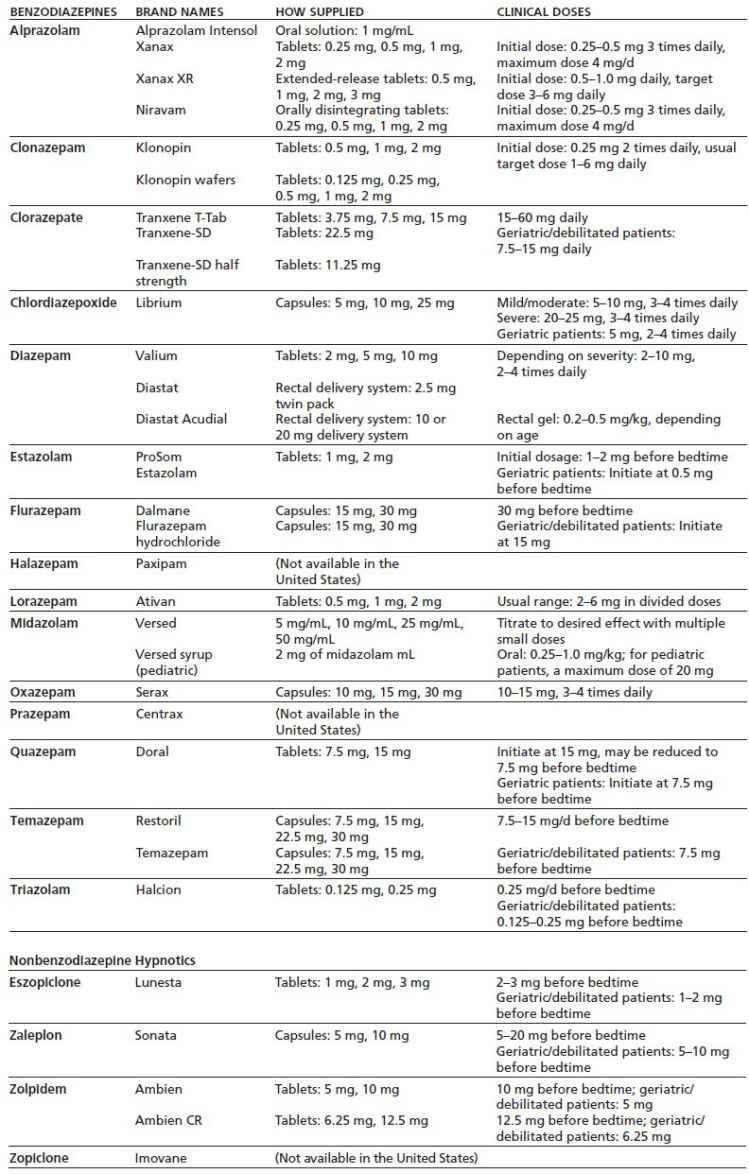
More recently, four nonbenzodiazepine hypnotics have been introduced: (a) zopiclone, a cyclopyrrolone; (b) eszopiclone, a stereoselective isomer of zopiclone; (c) zaleplon, a pyrazolopyrimidine; and (d) zolpidem, an imidazopyridine (4–6). Although these agents exert their hypnotic effects at the γ-aminobutyric acid (GABAA) receptor, their actions are not identical to classic benzodiazepines (see “Pharmacodynamics” section). The clinically available formulations of benzodiazepines and related drugs are shown in Table 8-1.
TABLE 8-1 FORMULATIONS OF BENZODIAZEPINES AND NONBENZODIAZEPINE HYPNOTICS
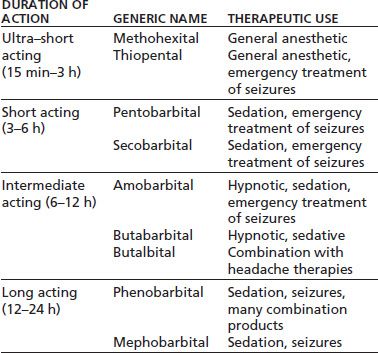
Barbiturates have largely been replaced by benzodiazepines because the latter drugs have greater safety and less potential for abuse; however, barbiturates continue to be used in general anesthesia and for the treatment of seizures (3,7,8). In the practice of addiction medicine, phenobarbital is used to treat difficult cases of sedative–hypnotic withdrawal, for example, in those cases unresponsive to benzodiazepines, detoxification from very high doses of sedative hypnotics, or when patients are abusing several different agents of this class. Specialists may also encounter patients who have been abusing analgesic preparations containing butalbital and require medically supervised detoxification. In these cases, heavy users may require phenobarbital withdrawal, whereas individuals taking less than or equal to 10 tablets per day of Fiorinal can usually be weaned off the medication gradually. Table 8-2 lists commonly used barbiturates (9).
TABLE 8-2 BARBITURATES
There are a number of other sedative hypnotics that are rarely seen in clinical practice. These include paraldehyde, chloral hydrate, and ethchlorvynol. The clinician may occasionally see meprobamate or, more commonly, the related drug carisoprodol, which is used as a muscle relaxant. All of these agents have abuse liability and produce a withdrawal syndrome upon abrupt discontinuation after chronic use. Phenobarbital withdrawal protocols are recommended for these drugs.
Barbituric acid was first prepared in 1864 by von Baeyer (7). Although not a central depressant itself, several of its derivatives have been used in medicine since the early 1900s. Barbital was introduced in 1903 and phenobarbital in 1912 (7). With respect to benzodiazepines, in the mid-1930s, Sternbach synthesized several heptoxdiazines, although it was not until 1955 when one of these quinazolines was treated with methylamine, that an active compound was developed (2). In 1957, the compound chlordiazepoxide was found to have hypnotic, sedative, and muscle-relaxing effects. The benzo-diazepines provided advantages over the older barbiturates because they were less toxic in overdose and had fewer drug interactions. In addition, they had superior efficacy and safety compared to meprobamate, which was introduced as a tranquilizer in 1955 (2). Nonbenzodiazepine hypnotics have been the most recent addition and may offer some advantages of lower abuse liability, though clinical experience is insufficient to make definitive judgments.
Benzodiazepines including alprazolam, lorazepam, and diazepam remain some of the most commonly prescribed psychiatric medications, with approximately 47.8 million prescriptions being written for alprazolam, the most frequently prescribed benzodiazepine, in 2011 (10). Estimates of the rates of abuse liability of benzodiazepines and related agents are made based on prescriptions issued, patterns of medical use, misuse by substance-abusing patients, and national surveys. Past year prevalence for the nonmedical use of sedative and tranquilizers has been reported to be 2.3% for adults in the United States with 9.8% of these individuals meeting criteria for abuse or dependence (11). There were 408,021 visits to emergency departments that involved benzodiazepine misuse or abuse in 2010 (12). This high number of visits is part of a trend noted since 1998 of increasing numbers of individuals seeking treatment for problems related to benzodiazepine abuse (13). Rates of emergency department visits in 2010 were 38.6 per 100,000 population in the United States for problems related to ben-zodiazepine use or misuse in patients 20 years or younger, the highest rate for any class of pharmaceutical agents (12). This rate was 168.8 visits per 100,000 population for persons aged 21 or older. This rate was only exceeded by the class of narcotic pain relievers.
Benzodiazepines comprised the second class of drugs most frequently involved in drug-related suicide attempts, with pain relievers being the class of drugs most often associated with these attempts (14). Overall, in 2009, benzo-diazepines accounted for 28.7% of visits for drug-related suicide attempts with 11.7% involving alprazolam and 8.1% related to the administration of clonazepam. The hypnotic agent zolpidem was involved in 5.5% of suicide attempt–related visits, while barbiturates, once an important class of drugs implicated in suicide attempts, only accounted for 0.8% of these visits.
Longer duration of treatment with benzodiazepines or the administration of higher doses of these agents may increase the odds for the development of dependence on these agents (15). Other factors that may increase the odds for the development of benzodiazepine dependence in older adults include being female, cognitive impairment, panic disorder, or the presence of suicidal thoughts (16). About 30% of psychiatric patients received benzodiazepines in one study, with the greatest use in patients with affective disorders, long duration of illness, and high users of psychiatric services (17). In general, the treatment of anxiety with medications has increased in recent years; however, the proportion of patients treated with antidepressants has grown, while the percentage of those treated with anx-iolytics has fallen slightly (18). In medical settings, most patients treated with benzodiazepines for anxiety do not increase their dose on their own. In fact, most patients tend to decrease the dose over time.
There are certain groups of high-risk patients where long-term use, misuse, and abuse are greater than in patients with anxiety disorders. Individuals with a history of alcohol abuse disorders comprise one group of individuals who have high rates of benzodiazepine abuse–related problems. In 2009, there were 109,192 emergency department visits related to the concurrent use of benzodiazepine and alcohol (14). Approximately 15% to 20% of alcoholic patients presenting for treatment may be abusing benzodiazepines (1,19,20). Several groups report a positive mood enhancement from acute benzodiazepine doses, including moderate and heavy drinkers (21,22), abstinent alcoholics (23–25), and individuals who have strong family histories of alcoholism (26–28). These findings suggest that mood elevation may be one of the factors contributing to use of benzodiaz-epines in alcoholic individuals and possibly to those with a family history of alcoholism.
Another major group of individuals for whom problems of benzodiazepine abuse are particularly high in both the United States and Europe are those with a history of opioid dependence. This includes both individuals who are actively abusing opioids and those who are receiving opioid substitution maintenance treatment (13). In methadone clinics, rates of urines positive for benzodiazepines are common, with 30% to 90% reporting illicit use (29,30). In a recent report, 47% of patients in a methadone maintenance program reported having a history of benzodiazepine use, with more than half of these patients starting the use of benzodiazepines after entering the methadone program (31). Rates of concurrent ben-zodiazepine use are also high for buprenorphine maintenance patients (32,33).
Pathways that mediate the biotransformation of the benzodiazepines are presented in Figure 8-2.
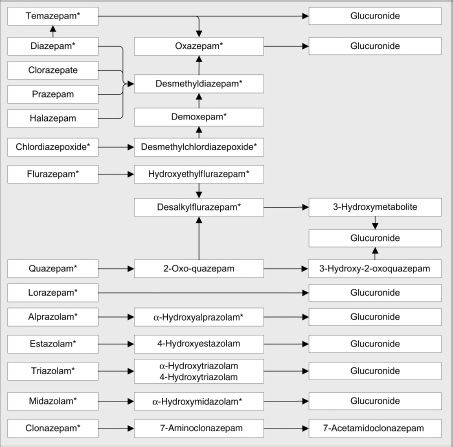
FIGURE 8-2 Biotransformation of benzodiazepines.
Many benzodiazepines undergo hepatic metabolism involving oxidative reactions mediated by the cytochrome P450 (CYP450) enzymes (47–49). Oxidative metabolism reactions include N-dealkylation or aliphatic hydroxylation. The CYP enzymes involved in the metabolism of benzodiazepines are listed in Table 8-3. The CYP3A4 enzyme mediates the oxidative metabolism of many of the benzodiazepines and also plays a role in the biotransformation of the non-benzodiazepine sedative–hypnotic agents, eszopiclone, zaleplon, and zolpidem. Several of the benzodiazepines are converted into active metabolites such as desmethyldiazepam that are very slowly cleared from the body. The final phase of metabolism for most benzodiazepines consists of conjugation of either the parent drug or their metabolites with glucuronide. Drugs or metabolites that undergo glucuronidation contain a hydroxyl group. Parent drugs, such as lorazepam and oxazepam, which undergo direct glucuronidation, are less subject to drug interactions or reduced clearance associated with impairment of hepatic function than are the other benzodiazepines.
TABLE 8-3 ELIMINATION HALF-LIFE (t1/2), CYTOCHROME P450 ENZYMES THAT MEDIATE METABOLISM, AND ACTIVE METABOLITES FOR BENZODIAZEPINES AND “Z” DRUGS
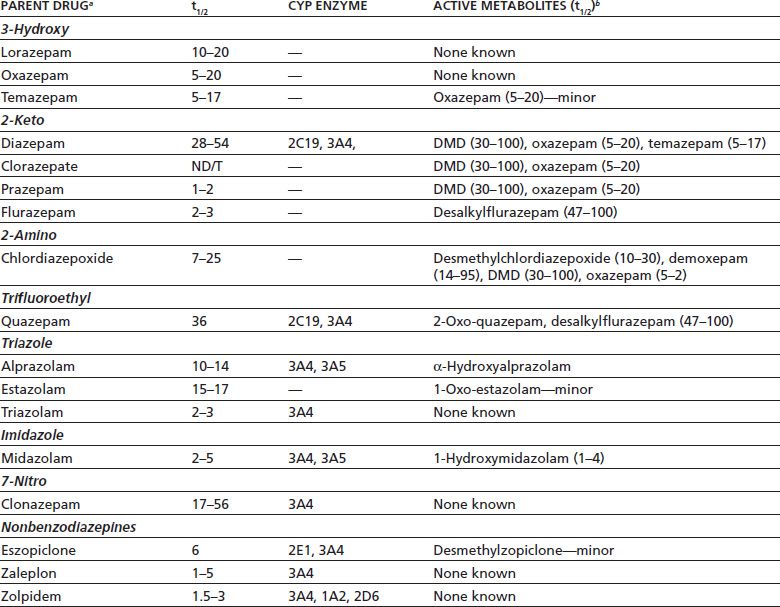
aBenzodiazepines are grouped by chemical class (bold).
bIf known; ND/T, not detectable or trace amounts detected in plasma; DMD, desmethyldiazepam; minor, very low metabolite concentrations detected.
Distinct metabolic pathways appear to mediate the biotransformation of the different Z drugs. Zolpidem is extensively metabolized by CYP3A4, CYP2C9, and CYP29 to inactive hydroxylated metabolites (34,35). Aldehyde dehydrogenase may play a major role in the metabolism of zaleplon, which involves the biotransformation of this drug into metabolites including 5-oxozaleplon that are excreted into the urine. Nearly 50% of zopiclone is transformed by esterase into a decarboxylated metabolite that is excreted through the lungs. This drug is also converted by CYP3A4 into the active metabolite zopiclone N-oxide and the inactive metabolite N-desmethylzopiclone.
The relationship between the pharmacokinetic profile of benzodiazepines and abuse liability is complex. It is generally believed that rapid onset of action is associated with euphoria (36). Onset of action after oral administration relies on the formulation of the drug, the intrinsic activity of the drug, lipid solubility, protein binding, and rate of entry into the brain. Some animal data suggest that greater lipid solubility increases the rate of brain uptake, with diazepam having more rapid brain uptake as compared to lorazepam (37). In clinical practice, pharmacokinetic factors do not always predict abuse liability. For example, clorazepate is rapidly decarboxylated in the stomach to desmethyldiaze-pam, which then reaches maximum plasma concentrations within 30 minutes or less (38), yet it is rarely cited as a benzodiazepine with high abuse potential. Subjective ratings by drug abusers of the “high” induced by clorazepate are lower than ratings of diazepam or lorazepam (39). Also, even though alprazolam and oxazepam differ only slightly in lipid solubility (40–42), intrinsic activity and rate of absorption are greater with alprazolam, which has higher abuse potential than oxazepam (23,24,43–46). Lower abuse potential is more consistently predicted with prodrugs that require hepatic metabolism to form the active moiety, such as the formation of desmethyldiazepam from halazepam, which appears to have lower abuse liability than diazepam (47). Therefore, the rate of absorption and entry into the brain may influence abuse liability, but other factors are also involved.
Benzodiazepines and related benzodiazepines receptor ago-nists such as eszopiclone, zaleplon, and zolpidem exert their clinical effects through allosteric modulation of the GABAA receptor (48). As GABA is the major inhibitory neurotransmitter system in the brain, positive modulation of the receptor by benzodiazepines is responsible for sedative, anticonvulsant, hypnotic, and amnestic effects of the drug. The GABAA receptor is a heteropentameric protein structure surrounding a central chloride channel (49). These receptors may be activated directly by agonists such as muscimol, which opens the central channel, in most cases leading to an influx of chloride ions, or indirectly by drugs such as benzodiazepines, which enhance the binding of GABA to the receptor and result in increased frequency of the opening of the central chloride channel (50). Both mechanisms lead to enhanced inhibitory effect by GABA neurons.
The five subunits that are linked to form the chloride ion channel are classified into several subtypes: alpha (α), beta (β), gamma (γ), delta (δ), epsilon (ε), rho (ρ), and pi (π), each of which has a unique sequence of amino acids (49). Minor alterations in amino acid sequences produce additional subtypes, which are designated by a number, and are relevant to the pharmacodynamic effects of drugs. Although the number of subunits, subtypes, and combinations may at first appear overwhelming, in reality, only a relatively small number of combinations are of importance in humans. In the human brain, the most common structure of the GABAA receptor consists of two α, two β, and one γ subunit (49). Benzodiazepines bind at the interface of the γ2 and α subunits (48).
In humans, α1 units have been identified and appear to be distributed in different regions of the brain, with the α1 unit located in the frontal, cortex, and other cortical areas, globus pallidus, hippocampus substantia nigra, and cerebellum (51). GABAA receptors containing the α5 subunit are prominent in limbic areas (52). Receptors containing α1, α2, α3, and α5 subunits are sensitive to the effects of benzodiazepines. The α5 subunit is located extrasynaptically and regulates tonic GABAergic currents, while the other subunits are located predominately within the synapse and mediate the rapid-phasic GABA currents (53). In contrast, the presence of α4 or α6 subunits leads to a lack of sensitivity to benzodiazepines.
GABAA receptors that contain α1 subunits appear to mediate the sedative–hypnotic effects of benzodiazepines (54–56). This subunit has also been implicated in playing a key role in the production of the ataxic effects of both benzodiazepines and zolpidem (55,57). The antianxiety effects of benzodiazepines may be mediated by receptors containing the α2 and possibly the α3 subunit (55,58). The α2 and α3 subunits also may be involved in the mediation of the muscle relaxant effects of benzodiazepines (59). There is also evidence that the α2 and α3 subunits may mediate the rewarding effects of diazepam (60). Other research, in contrast, has implicated the α1 in mediating the reinforcing effects of benzodiazepines (61). Limited findings suggest that the α5 subunit is associated with sedation, learning, and memory in animal models (57,62).
Barbiturates share some pharmacodynamic properties with the benzodiazepines. At low concentrations, barbiturates act as positive modulators of the GABAA receptor via an allosteric mechanism (63). However, at higher concentrations, barbiturates act as direct GABAA receptor agonists by prolonging the duration of the opening of the chloride channel. GABAA receptors that contain the β2 or β3 subunit show greater sensitivity to the effects of pentobarbital than receptors with the β1 subunit (58). Pentobarbital shows the greatest efficacy as a GABA receptor agonist in receptor complexes that contain the α6 subunit, which is a benzodiazepine-insensitive subunit (64). Barbiturates may reduce excitatory neurotransmission through inhibition of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and inhibit neurotransmitter release by blockade of voltage-sensitive calcium channels (65–67).
The Z drugs have been identified that act as positive modulators of the effects of GABA agonists on the GABAA receptor. As would be expected, these drugs share many of the pharmacologic actions with the classic benzodiazepine agents including sedative–hypnotic, anxiolytic, myorelaxant, and anticonvulsant effects, although their selectivity for these actions differs. For example, animal studies suggest that zolpidem is more selective in its sedative effects as compared to its anticonvulsant actions than are quazepam, zaleplon, or zopiclone (68,69). Among the Z drugs, eszopiclone has the strongest antianxiety effect in animal models, which is produced at nonhypnotic doses (70,71). The Z drugs may differ from the classic benzodiazepines in that their amnestic effects may be less pronounced and tolerance is less likely to develop to their pharmacologic actions (4).
Z drugs and benzodiazepines may act through overlapping binding sites on the interface between α and β subunits of the GABAA receptor. This is suggested by findings that the Z drugs inhibit the binding of the benzodiazepine flunitrazepam to brain tissue (6,72,73). Also, the enhancement of muscimol binding produced by zaleplon is blocked by the administration of the benzodiazepine receptor antagonist flumazenil. Studies involving the selective replacement of the amino acids that comprise the GABAA receptor subunits indicate that zolpidem and zopiclone interact with amino acids located on specific sites on the α and β subunits that also interact with benzodiazepines (74–76). These studies have also demonstrated that zolpidem, however, binds to sites on these subunits that are not crucial for benzodiazepine activity.
Dissimilarities in the activity between individual Z drugs or between Z drugs and the classic benzodiazepines may arise, in part because of differences in the affinity of these drugs for the different subtypes of GABAA receptors. Benzodiazepines have roughly similar affinities for GABAA subtype receptors that contain the α1, α2, α3, and α5 subunits. In contrast, zaleplon and zolpidem have more than 10-fold greater affinity for receptors with a α1β2γ2 composition than for those with a α2β2γ2 composition, whereas zopiclone has essentially equivalent affinity for these receptors (77). Zolpidem in contrast to the benzodiazepines and other Z drugs has little affinity for α5-containing GABAA receptors, and the presence of the γ3 subunit produces insensitivity to zolpidem (77).
Differences in binding affinities for the different GABAA receptor subtypes roughly correlate with the potency of the positive GABAA receptor modulators to enhance the GABA-mediated currents (78). The full functional implications of differences in the affinities of Z drugs for the different GABAA receptor subtypes, however, remain far from being clear. A 10-fold difference in affinity for different receptor subtype may not be great enough to result in a distinct pharmacologic profile for a drug. There is some evidence, however, that zolpi-dem may have pharmacologic actions that are associated with a selective action on GABAA receptors that contain the α1 subunit. In genetically altered mice that are missing the gene for the GABAA α1 subunit, all of the high-affinity binding for zolpidem in the cortex is lost (79). In these mice, the hypnotic effects of zolpidem are greatly diminished, whereas those of diazepam are increased, which is consistent with a selective action of zolpidem on the α1-containing receptors (80).
The most serious drug–drug interactions occur when sedative hypnotics are combined with alcohol or other drugs that depress CNS activity. Benzodiazepines do not induce their own metabolism; however, those that are metabolized through CYP3A4 (see Table 8-3 ) are subject to altered plasma levels by agents that inhibit this metabolic pathway. Common inhibitors are ketoconazole, itraconazole, mac-rolide antibiotics (erythromycin), fluoxetine, nefazodone, and cimetidine. Drugs that induce or inhibit CYP2C19 may also influence the metabolism of some benzodiazepines. For example, oral contraceptives containing estrogen and progesterone impair the metabolism of some substrates of CYP1A2, CYP3A4, and CYP2C19, although findings with benzodiaz-epines are inconsistent (81). One study found inhibition of alprazolam metabolism in women taking low-dose estrogen oral contraceptives (82), and another did not (83). Similarly, lorazepam metabolism was unaffected by oral contraceptives in one study (84), yet increased in another (82). The latter study also found enhanced elimination of temazepam. Oxazepam kinetics are not affected by oral contraceptives (84). The clearances of both chlordiazepoxide and diazepam are impaired in women taking oral contraceptives (85–87).
Inducers of P450 enzymes, for example, rifampin, may significantly reduce plasma concentrations of zaleplon, zolpidem, and zopiclone (34). Similarly, repeated administration of the CYP3A4 inducer carbamazepine increases the clearance of zolpidem (88). Administration of the CYP3A4 enzyme inhibitors such as erythromycin or ketoconazole, in contrast, can decrease the clearance of zolpidem (34).
Barbiturates present a serious risk of CNS depression, coma, and death when taken in high doses or with ethanol or other sedative hypnotics. They induce their own metabolism (pharmacokinetic tolerance) and induce CYP2B6, CYP2C9, and CYP3A4, resulting in enhanced metabolism of drugs that are substrates of these cytochromes, reducing their therapeutic effects. Patients taking phenobarbital may experience decreased effects of anticoagulants, oral contraceptives, corticosteroids, some antibiotics, and other drugs (89).
As the abuse of prescription opioids has increased, the interaction between these drugs and sedative hypnotics has become an increasing concern. There is extensive evidence implicating benzodiazepines in both fatal and nonfatal cases of opioid overdose (13). Pharmacodynamic interactions between benzodiazepine and opioids may lead to oversedation and impaired motor performance. The combination of oxycodone and alprazolam, for example, when coadminis-tered at therapeutic doses, produced greater impairment in psychomotor performance or increased difficulty in concentrating than was seen when these agents were administered alone (90). High-dose diazepam also enhanced psychomotor impairment and sedation when given concurrently with either buprenorphine or methadone (91).
Although the paths to sedative–hypnotic addiction are complex and vary among individuals, it is helpful to consider three characteristics of the drug class that are related to misuse: (a) hedonic effects, (b) tolerance, and (c) the withdrawal syndrome. Use of benzodiazepines purely for the hedonic value, that is, to achieve a pleasurable or euphoric mood change, is rare unless they are used in combination with other drugs such as methadone, which results in a “boost.” Benzodiazepines with a rapid onset of action, such as alprazolam or diazepam, probably present the greatest risk for this type of abuse.
Tolerance of clinical effects may lead some patients to escalate the dosage. The risk appears greatest when the drugs are used as hypnotics because tolerance of sedation occurs rapidly, though there is some evidence that temazepam (92) and eszopiclone (93,94) have lower risk of tolerance than other agents. There is also evidence that tolerance does not develop to the anxiolytic action of benzodiazepines (95), and patients treated for panic disorder are more likely to decrease rather than increase their benzodiazepine dose; however, the issue of tolerance to the antianxiety effect remains controversial (96). There are also some reports of patients who have improvement in anxiety symptoms after discontinuation of long-term benzodiazepine treatment, suggesting that for some individuals, chronic treatment with these drugs actually exacerbates symptoms (97). Tolerance of the antiepileptic effects is well recognized and limits the use of these agents for long-term treatment of seizures, though the time to develop tolerance differs among drugs within the class (98). Partial tolerance develops to several other actions of the benzodiazepines, such as psychomotor and cognitive impairment (99–101), which may permit some patients to gradually escalate doses to supratherapeutic levels.
The withdrawal syndrome that appears upon decreasing dosage or abrupt discontinuation of treatment may produce uncomfortable mental and physical states that make it difficult for patients to terminate drug use. Rebound insomnia is a particular problem for patients who discontinue benzodiazepines. The withdrawal syndrome is discussed in greater detail later in this chapter.
The mechanism of therapeutic action is well known for benzodiazepines and barbiturates; the neurophysiologic basis of their reinforcing effects is not well understood. Recent findings, however, suggest that benzodiazepine may increase the firing of dopaminergic neurons within the ventral tegmental area (VTA), an effect that may activate the mesolimbic reward systems implicated in mediating the reinforcing effects of many commonly abused drugs (61). This action may result from the inhibition of interneurons within the VTA that function to suppress the activity of dopaminergic neurons within this structure and may involve GABAA receptors containing the α1 subunit.
The pharmacodynamic mechanisms underlying the development of tolerance and withdrawal to sedative hypnotics have been extensively studied. Tolerance, as evidenced by decreased responsiveness of GABAA receptors to benzodiazepines, has been demonstrated using measures of both electrophysiologic activity (102,103) and GABA-mediated chloride flux (104). The decreased ability of benzodiazepines to positively modulate GABAA receptors may result, in part, from the loss of interaction between benzodiazepine- and GABA-binding sites within these receptors, referred to as “uncoupling” (105,106). Zolpidem may also produce uncoupling between these binding sites (107). Another possible mechanism of tolerance may involve benzodiazepine-induced internalization of surface GABAA receptors into intraneuronal sequestration sites (106). Alterations in sensitivities to the effects of benzodiazepines might also result from changes in the subunit composition of brain GABAA receptors. In vitro studies indicate that levels of the messenger RNAs (mRNAs) that are involved in the synthesis of the α1 GABAA receptor subunit are reduced during prolonged exposure to benzodiazepines, whereas mRNAs for the benzodiazepine-insensitive α4 subunit are increased soon after benzodiazepine withdrawal (108). In animals, tolerance to benzodiazepines has occurred in association with decreases in the density of binding sites for hippocampal GABAA receptors that contain the α5 subunit (105). After withdrawal from the neurosteroid precursor, progesterone, α4 subunit mRNAs, and protein levels are increased in the brain, as may occur during withdrawal from benzodiazepines (109). These changes in α4 subunit expression correlate with decreased sensitivity to the effects of benzodiazepines on GABA-mediated chloride currents (109). This loss of sensitivity can be prevented by blockade of the expression of α4 subunits. These findings point to the possible role of increased α4 subunit expression in early benzodiazepine withdrawal.
The glutamatergic system may play a major role in the benzodiazepine withdrawal syndrome. In animal models, anxiety associated with benzodiazepine withdrawal may involve up-regulation of hippocampal AMPA receptors as evidenced by increased AMPA receptor GluR1 subunits and increased AMPA receptor binding (110–113). Other studies have confirmed increased AMPA receptor binding in the hippocampus and thalamus using different experimental paradigms of withdrawal but have not found corresponding alterations in levels of the GluR1 or GluR2 subunits in these brain regions (114). AMPA-mediated conductances in hippocampal neurons have been shown to be enhanced by abrupt withdrawal from benzodiazepines (115). These changes occur in association with the presence of anxiety-related behaviors.
The benzodiazepines occupy an intermediate position of abuse liability, with barbiturates and older sedative hypnotics (e.g., methaqualone, ethchlorvynol) having greater risk of abuse, whereas anxiolytics and hypnotics that act via non-GABAergic mechanisms (e.g., buspirone, antidepressants, ramelteon) lack abuse potential. Many authorities believe that zolpidem, zopiclone, eszopiclone, and zaleplon have lower potential for abuse than classic benzodiazepines; however, there are case reports of abuse and abstinence syndromes associated with abrupt discontinuation of high doses after prolonged use of these agents. Also, laboratory studies have shown that zolpidem administration increases subjective responses such as “drug liking” and “good effects” in both experienced drug users (116) and healthy subjects (117), suggesting that this agent does have abuse potential. More controversial is the issue of relative abuse liability among the benzodiazepines themselves. Using the sole criterion of a positive mood effect in humans, the benzodiazepines with the highest liability for abuse are flunitrazepam, diazepam, alprazolam, and possibly lorazepam. Those with the lowest positive reinforcing effects in humans are clonazepam, chlordiazepoxide, halazepam (47), prazepam (118), quazepam, and oxazepam. That said, individuals at risk for sedative–hypnotic abuse (e.g., alcoholics) may misuse any of the benzodiazepines, even those with relatively low potential for abuse. Benzodiazepines with the highest abuse potential produce a rapid onset of pleasant mood, well-being, relief of dysphoria, a sense of increased popularity, the belief that thoughts flow more easily, and a general state of contentment (119).
The relative abuse liability of the barbiturates and the Z drugs, compared to benzodiazepines, is a matter of some controversy. In human laboratory studies that assess abuse potential, these drugs produce euphoric effects at doses above their typical therapeutic ranges. For example, eszopiclone, at doses of 6 mg and 12 mg, produced euphoric effects comparable to 20 mg of diazepam in previous sedative–hypnotic abusers. Studies examining the abuse liability of 25 mg, 50 mg, and 75 mg of zaleplon, in subjects with a history of sedative–hypnotic abuse, indicated abuse potential similar to benzodiazepines and benzodiazepine-like hypnotics. Zolpidem, 10 mg, produced effects similar to 0.25 mg of triazolam (120), but higher doses of triazolam (0.75 mg) were more often rated as similar to barbiturates than high doses of zolpidem (45 mg). Furthermore, post-marketing surveillance indicates a relatively low potential for abuse for zolpidem considering how often it is prescribed (121).
TOXICITY STATES AND THEIR MEDICAL MANAGEMENT
Toxicity must be considered whenever therapeutic agents are prescribed. With respect to sedative hypnotics, benzodiazepines provide a greater margin of safety than barbiturates and older agents. Despite the improved safety profile, prescription of benzodiazepines is associated with acute and chronic risks.
In the therapeutic context, acute toxicity of benzodiazepines includes sedation, psychomotor impairment, and memory problems. It is well established that acute doses of benzodiazepines produce anterograde amnesia, difficulty acquiring new learning (122), and sedation that may affect attention and concentration. Tolerance usually develops to most of the cognitive effects, but not in all patients. Those who use intermittent doses (“p.r.n”) of high-potency agents may not develop tolerance and continue to be at risk for impaired psychomotor and memory function, especially in the first few hours after taking a dose. Furthermore, studies (123) and clinical experience suggest that not all benzodiazepines produce the same type or severity of cognitive impairment. Although many studies have found no cognitive impairment associated with long-term benzodiazepine treatment (124,125), others have reported persistent problems in psychomotor function, learning, concentration, and visuospatial skills (126,127). In chronic high-dose users, greater impairment is seen in men, elderly, and individuals taking the highest doses (122). A meta-analysis suggested a pervasive cognitive impairment in chronic users, but the interpretation is complicated by comparing studies with different methods and populations (126). Even in patients with persistent cognitive impairment detected by neuropsychological testing, there is substantial improvement when benzodiazepines are discontinued, though patients still perform worse than controls at 6 months after the medication is stopped (128). Most patients do not report persistent cognitive difficulties, either because they underestimate the impairment or because the neuropsychological test findings have little relevance for everyday life.
Anterograde amnesia occurs with therapeutic doses of benzodiazepines, and the impairment in learning new information represents a major drawback of these medications. Some tolerance develops to the cognitive impairment, but it is not complete. Controlled studies that take into account alcohol use, psychiatric disorders, and age indicate that a mild cognitive disturbance may persist but that it does not interfere with the demands of everyday function.
An exception may be the elderly patient who is particularly vulnerable to falls and memory problems, though one study found the memory impairment in elderly taking benzodiazepines was rather small (129). Falls, conversely, present a serious problem for the elderly. Classic benzodiazepines, nonbenzodiazepine hypnotics, SSRI antidepressants, and antipsychotics have all been linked to falls and fractures in the elderly (130–132), making the treatment of insomnia in aged patients a challenge.
Zolpidem also produces anterograde amnesia and has been associated with somnambulism and complex nocturnal behaviors, such as eating, shopping, and driving. Similar problems may be seen with zaleplon, especially at high doses. It is not known whether all hypnotics are capable of producing such effects; however, the U.S. Food and Drug Administration (FDA) issued a request for a label change requiring that benzodiazepine and nonbenzodiazepine hypnotics include a warning describing these complex sleep-related behaviors.
Several surveys in different countries have found that there is a higher incidence of motor vehicle accidents associated with benzodiazepines (133,134). It is not known whether this reflects acute psychomotor impairment, falling asleep at the wheel, or persistent visuospatial impairment. Zolpidem at a high (20 mg) dose when administered in the middle of the night can produce in decrements in driving performance in the morning (135).
The risks of benzodiazepines during pregnancy and lactation have been the subject of controversy. Data pooled from cohort studies have not demonstrated an increased risk of major malformations or cleft palate (136). Conversely, when data from case–control studies were pooled in the same meta-analysis, a small but statistically significant association was found between exposure to benzodiazepines and oral cleft abnormalities or other major malformations. The rate of cleft palate in the general population is estimated at 0.06% (137), and case–control studies show that with benzodiazepines, the risk may be approximately doubled, still rather low at 0.12%. A review of older studies assessing the possible association of benzodiazepine use in the first trimester and cleft palate found that the use of different sampling schemes, different benzodiazepines at different doses for different durations, and failure of some studies to account for concurrent medications or illnesses contributed to the “confusion and controversy regarding the safety of benzodiazepine use during pregnancy” (138).
In a well-designed study of 873,879 infants whose mothers were registered in the Swedish Medical Birth Register, 1,979 infants were exposed to benzodiazepines or nonbenzodiazepine hypnotics (mainly zopiclone), but increased risk of orofacial clefts was not found (139). They did find an increased risk for low birth weight in infants that had both early and late exposure to benzodiazepines in utero, but it was most evident in women who also had been taking an antidepressant. They also found an unexpectedly high number of infants with pylorostenosis and alimentary tract atresia, although concomitant exposure to antidepressants and an anticonvulsant complicates the interpretation of the finding. Another study found that infants exposed in the first trimester to the combination of serotonin reup-take inhibitor antidepressants and benzodiazepines had increased risk of congenital anomalies and congenital heart disease compared to unexposed; however, when maternal illness was controlled, only the increased risk for congenital heart disease remained significant (140). Monotherapy with either class of drug was not associated with an increased risk. Most recent studies do not find an association of in utero benzodiazepine exposure alone with major congenital anomalies (141,142).
Although the major concern of clinicians has been congenital anomalies associated with benzodiazepines, two other clinically important problems may be encountered during pregnancy. Newborns who have been exposed to benzodiazepines in utero during the last trimester or during delivery may present with floppy baby syndrome, which is characterized by low Apgar scores, poor sucking, hypotonia, poor reflexes, and apnea (143). Neonatal withdrawal syndromes have also been reported (144).
Benzodiazepines administered to nursing mothers enter the breast milk but appear in such low concentrations that they do not usually cause adverse effects in infants (145– 148). There are two important exceptions to this general rule: The risk to the infant is higher (a) if the benzodiaz-epine is given in high doses antepartum and continued postpartum and (b) if infants have impaired hepatic function, as evidenced by hyperbilirubinemia (147). Despite their relative safety, breast-fed infants whose mothers are taking benzodiazepines should be monitored for lethargy, weight loss, and signs of an abstinence syndrome.
The use of anticonvulsants, including phenobarbital, by pregnant women with epilepsy has been associated with reports of harm to the fetus, although the findings are not consistent (149). The frequency of anticonvulsant embryopathy, defined as major malformations, growth retardation, and hypoplasia of the midface and fingers, is increased in infants exposed to phenobarbital alone or in combination with other anticonvulsants, compared to those whose mothers had a history of epilepsy but took no medications during pregnancy (150).
All sedative hypnotics produce effects on a continuum from sedation to obtundation. Barbiturates have a greater risk for respiratory depression than do benzodiazepines. In overdose situations, sedative hypnotics are often combined with ethanol or other CNS depressants. When high doses of benzodiazepines are ingested, as either a therapeutic intervention or overdose, initial signs of toxicity are ataxia and impaired gag reflex. Rarely, sedative hypnotics produce disinhibition or paradoxical excitement.
A severe withdrawal syndrome after high-dose chronic administration of chlordiazepoxide or diazepam was demonstrated in the early 1960s (151,152) and, in its most severe form, can include grand mal seizures and psychosis. A characteristic abstinence syndrome may develop upon abrupt discontinuation of therapeutic doses of benzodiazepines that are administered for several weeks (153). When administered for short periods and at therapeutic doses, the withdrawal syndrome is usually mild, consisting of anxiety, headache, insomnia, dysphoria, tremor, and muscle twitching. After long-term treatment with therapeutic doses, the syndrome increases in severity and may include autonomic dysfunction, nausea, vomiting, depersonalization, derealization, delirium, hallucinations, illusions, agitation, and grand mal seizures. The time course of the abstinence syndrome is related to the half-life of the agent, with patients taking short–half-life agents (lorazepam, alprazolam, temazepam) developing symptoms within 24 hours of discontinuation, the severity of which peaks at 48 hours. With longer– half-life agents such as diazepam, symptoms may develop a week after drug discontinuation and last for several weeks. This timeline should be used as a general guideline because some patients on long-acting agents will develop symptoms earlier than predicted by the pharma-cokinetics of the drug. In addition, some clinicians believe that there is prolonged withdrawal syndrome that persists for several months, but it has not been clearly distinguished from return of original anxiety symptoms.
In general, longer treatment periods, higher doses, sudden drug discontinuation, and psychopathology increase the severity of the abstinence syndrome. Clinical experience has shown that there is great variability in the sensitivity of patients to discontinuation of benzodiazepines, and all patients who have been taking the drug for several weeks or longer should have the medication tapered.
The withdrawal syndrome from barbiturates was described by Wikler (154) using 0.8 to 2.2 g/d oral doses of secobarbital or pentobarbital for 6 weeks or longer. Upon abrupt discontinuation, apprehension, uneasiness, muscular weakness, coarse tremors, postural hypotension, anorexia, vomiting, and myoclonic jerks occurred within the first day and lasted up to 2 weeks. Grand mal seizure occurred within 2 to 3 days of discontinuation and lasted as long as 8 days. Delirium was most likely to develop 3 to 8 days after drug discontinuation and lasted up to 2 weeks. Strategies for management of the barbiturate withdrawal syndrome include transition to an equivalent dose of phenobarbital, determined by either a challenge dose or loading dose procedure (155).
Benzodiazepines are the most widely used and abused drugs of e sedative–hypnotic class. In animal models and human laboratory studies, they occupy an intermediate position of abuse liability that is lower than barbiturates, but higher than anxiolytics such as buspirone (23) that do not act at the GABAA receptor complex. In patients with anxiety disorders, abuse is not common; however, certain subgroups of patients, such as individuals with alcohol dependence and those in methadone maintenance programs, are at high risk to misuse these agents. Compared to the general population, higher rates of benzodiazepine use are found in the elderly and patients with chronic pain; however, there is insufficient evidence to suggest these groups abuse benzodiazepines. The newer non-benzodiazepine hypnotics, commonly referred to as the Z drugs, may have a lower potential for abuse, tolerance, and dependence, though they are not devoid of such risk. The identification of GABAA receptor subtypes and clarification of their function provide hope that drug development will lead to GABAA agonists and modulators that have fewer adverse effects, lower risk for dependence, and greater specificity of action (156).

Among the substances termed “club drugs” by the National Institute on Drug Abuse is γ-hydroxybutyrate (GHB). A sedative–hypnotic, it is a simple, four-carbon molecule synthesized in the early 1960s in a search for an orally active γ-aminobutyric acid (GABA) analog. It has been used as a general anesthetic and to induce absence seizures in animal models of epilepsy. More recently, it has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of cataplexy in narcolepsy (marketed as Xyrem) and is used for treating alcohol and opiate dependence primarily in Italy and Austria, where it is marketed as Alcover (1).
GHB abuse reports date to the early 1990s. Initially available as a dietary supplement in the United States, bodybuilders were attracted by reports that GHB raised levels of growth hormone. The substance is taken orally, with mis-users reporting a euphoric “high” from the drug; the effects have been described anecdotally as comparable to both alcohol intoxication (with disinhibition, drowsiness, and loss of motor control) and MDMA/Ecstasy use (enhanced sensuality, empathogenesis) (2). In the United States, GHB is a Schedule I controlled substance, except that the FDA-approved formulation (for narcolepsy) is classified as Schedule III. Other countries maintain similar regulatory classifications.
GHB is inexpensive and readily available at some social events (notably dance clubs), or via kits and recipes for home manufacture found on the Internet. Other names for GHB include “G,” “liquid ecstasy,” “Fantasy,” “Grievous Bodily Harm,” “Cherry Meth, “soap,” “salty water,” and “Georgia Home Boy” (3). Street names that imply that GHB is a stimulant, for example, liquid ecstasy and Cherry Meth, which may lead to underestimation of its ability to cause physical dependence. GHB is usually sold in a salty-tasting, aqueous solution that may have a variable concentration, undoubtedly contributing to the high incidence of adverse effects. It is also sold as a white powder ready for dissolution. Legal restrictions on the purchase of GHB have led to increased abuse of two GHB prodrugs: γ-butyrolactone (GBL), whose street names include “lactone,” “Renewtrient,” “Blue Nitro,” and “Verve”; and 1,4-butanediol (BD), also known as “Pro-G,” “Thunder,” “Weight Belt Cleaner,” and “Pine Needle Extract.” Both substances are available as industrial solvents. GBL is a Drug Enforcement Administration List 1 chemical in the United States, requiring justification for sales, and the FDA classifies BD as a Class I Health Hazard (i.e., a potentially life-threatening drug).
PHYSIOLOGY AND PHARMACOKINETICS
Found endogenously at very low concentrations in the brain, GHB is both a precursor and degradation product of GABA and is structurally similar to GABA. However, unlike GABA, GHB readily passes the blood–brain barrier. GHB interacts in a complex way with endogenous GABA (the primary inhibitory neurotransmitter in the brain) and with GABA receptors (4). Oral doses are rapidly absorbed, with maximum plasma concentrations reached in 30 to 50 minutes. Plasma elimination half-life is in the range of 20 to 30 minutes (5,6). GHB is metabolized to succinate, and then further to CO2 and water via the citric acid cycle. GHB binds to and activates two receptors. It has high affinity for excitatory GHB receptors and low affinity for the inhibitory GABAb receptor. Human GHB receptors have recently been cloned and are pharmacologically distinct from GABA receptors—they are G-protein coupled and do not bind GABA (7). GHB affects dopamine release biphasically—low GHB concentrations stimulate dopamine release via the GHB receptor (which may increase addiction liability by activating reward pathways), while higher concentrations inhibit dopamine release via the GABA receptors. So, after an initial inhibitory phase, as concentration of GHB in the brain decreases, dopamine release increases due to activation of GHB receptors. This results in both a sedative and stimulatory effect in recreational users and also causes sudden awakening after several hours of GHB-induced deep sleep (8). GHB also influences serotonin turnover and affects neurosteroid, acetylcholine, and growth hormone levels (4).
The pharmacologic profile of BD is similar to that of GHB (9). GBL has greater lipid solubility, is more rapidly absorbed, and reaches a higher maximum plasma concentration of GHB than an equimolar dose of GHB (10,11). Recent animal studies indicate that both BD and GBL are more potent and longer acting than GHB (12). GHB can be detected for approximately 5 hours in blood and oral fluid and less than 12 hours in urine (13).
EPIDEMIOLOGY
Estimates vary on the frequency of GHB use worldwide. In the United States, there are limited data on the use of GHB by adults since GHB was only added to the U.S. National Survey on Drug Use and Health in 2006 (1). The Monitoring the Future Study estimated that the annual rates of GHB use by 8th, 10th, and 12th grade students are low (0.7% to 1.1%) and have declined from 2000 (the year in which GHB was added to the survey) to 2009 (14). However, sizable minorities in some populations report higher use rates. For example, 7% of young adults and adolescents in treatment for substance abuse reported lifetime use of GHB (15). A study of 450 “club” drug-using gay and bisexual men in New York City found that 29% reported GHB use in the previous 4 months (16). Although European data suggest that the use of GHB is generally low, GHB abuse seems to be gaining popularity in some parts of Europe and Australia (8).
Intoxication
Reasons for GHB use include “to be sociable,” to enhance sex, and to explore altered states of consciousness. Subjective effects of GHB include slurred speech, ease in socializing, feelings of increased sexual intimacy, drowsiness, and feelings of depression after the GHB “high” is over (17). GHB administered under controlled laboratory conditions produces sleepiness, sedation, fatigue, and feelings of being “easy going” or “mellow.” Effects noted by observers of intoxicated subjects include decreased psychomotor performance and level of alertness and increased ratings of muscle relaxation and abnormal posture (18). In overdose, clinical characteristics include those expected from a central nervous system (CNS) depressant—bradycardia, vomiting, somnolence, obtundation, stupor, and coma—but a literature review also identified agitation, combativeness, and self-injurious behavior as relatively common in persons using GHB alone and with cointoxicants (19). The frequent use of alcohol with GHB is thought to worsen GHB-induced sedation and may increase episodes of vomiting, hypotension, and respiratory depression (9). Simultaneous use of alcohol and other drugs in more than half of all patients with GHB intoxication causes atypical symptoms and impedes correct diagnosis of GHB intoxication (8).
Abuse Liability, Dependence, and Withdrawal
Physical dependence and addiction have been reported with GHB, GBL, and BD (20,21). Knowledge of dependence on these drugs comes from numerous case reports and small case series; controlled withdrawal studies have not been conducted. One estimate places the likelihood for GHB abuse intermediate to triazolam and pentobarbital (18). Dependence may develop rapidly, usually with frequent (four or more times a day) dosing. Time course estimates for the development of physical dependence and severe withdrawal range from 7 days (22) to use over 2 months or more (21,23,24).
Although the withdrawal syndrome may be mild, with insomnia, agitation, anxiety, and limited sympathetic arousal, an analysis of reports of GHB withdrawal found that severe, potentially life-threatening withdrawal states may develop requiring vigorous inpatient clinical management. Signs and symptoms are similar to those seen in alcohol withdrawal: tremor, tachycardia, restlessness, and delirium, including hallucinations (25).
Adverse Effects
Because the dose–response curve for GHB is steep and concentrations of GHB in illicitly purchased or homemade solutions are notoriously hard to predict, overdose is a hazard. Symptoms of CNS depression are dose related, with reports of somnolence within 15 minutes at a dose of 30 mg/kg, and unconsciousness and coma at doses exceeding 50 mg/kg (26). Nonfatal overdose may lead to intubation and management in an ICU, although in one study the majority of overdose patients were discharged within 6 hours of presentation (27). There are limited data on the long-term sequelae of use of GHB, BD, and GBL. Animal models suggest that GHB causes lasting deficits in memory and social interaction and increases in anxiety due to long-term neuroadaptations in the brain oxytocin systems (28).
GHB-associated deaths, both with and without cointoxicants, have been identified in the United States, Europe, and Australasia (29). GHB use is associated with risky behaviors such as coingestion of ethanol, driving under the influence of GHB (30), and risky sexual behaviors (31). GHB has been associated with drug-facilitated sexual assaults (32,33). Such use may be facilitated by GHB’s properties of being colorless, odorless, sedating, purportedly causing amnesia, and poorly detected in urine after 12 hours.
Treatment
McDonough, Kennedy et al. conducted a retrospective analysis of treatment for GHB withdrawal and gave recommendations for management. In their review, a tapering regimen of benzodiazepines was used in 91% of 38 cases, with a mean dose in diazepam equivalents of 335 mg (range 20 to 2,655 mg). The mean duration of withdrawal was 9 days. In 82% of the 38 cases, other drugs were used in combination with benzodiazepines—anti-psychotics for psychosis and delirium, as well as anti-convulsants and nonbenzodiazepine sedatives such as pentobarbital. They note that withdrawal delirium developed in more than half the cases where GHB use occurred every 8 hours or less, or with use of more than 30 g of GHB a day. They recommend inpatient management of withdrawal in these cases, and note that while antipsychotics have been used for delirium and psychosis, they do not appear sufficient or necessary. Pentobarbital was effective where symptoms of withdrawal persisted despite high doses of benzodiazepines. They conclude by noting that management of GHB withdrawal requires more study (25). More recently, a 1-week inpatient tapering schedule of GHB has been shown to suppress GHB withdrawal; this may be a reasonable treatment approach in cases requiring hospitalization, if GHB is available (34).
REFERENCES
1. Carter LP, et al. Illicit gamma-hydroxybutyrate (GHB) and pharmaceutical sodium oxybate (Xyrem): differences in characteristics and misuse. Drug Alcohol Depend 2009;104(1-2):1–10.
2. Galloway GP, et al. Abuse and therapeutic potential of gamma-hydroxybutyric acid. Alcohol 2000;20(3):263–269.
3. Maxwell JC. Party drugs: properties, prevalence, patterns, and problems. Subst Use Misuse 2005;40(9-10):1203–1240.
4. Drasbek KR, Christensen J, Jensen K. Gamma-hydroxybutyrate—a drug of abuse. Acta Neurol Scand 2006;114(3):145–156.
5. Palatini P, et al. Dose-dependent absorption and elimination of gamma-hydroxybutyric acid in healthy volunteers. Eur J Clin Pharmacol 1993;45:353–356.
6. Brenneisen R, et al. Pharmacokinetics and excretion of gamma-hydroxybutyrate (GHB) in healthy subjects. J Anal Toxicol 2004;28(8):625–630.
7. Andriamampandry C, et al. Cloning and functional characterization of a gamma-hydroxybutyrate receptor identified in the human brain. FASEB J 2007;21(3):885–895.
8. van Amsterdam JG, et al. Risk assessment of gamma-hydroxybutyric acid (GHB) in the Netherlands. Regul Toxicol Pharmacol 2012;63(1):55–63.
9. Thai D, et al. Clinical pharmacology of 1,4-butanediol and gamma-hydroxybutyrate after oral 1,4-butanediol administration to healthy volunteers. Clin Pharmacol Ther 2007;81(2):178–184.
10. Kohrs FP, Porter WH. Gamma-Hydroxybutyrate intoxication and overdose. Ann Emerg Med 1999;33(4):475–476.
11. Wood DM, Brailsford AD, Dargan PI. Acute toxicity and withdrawal syndromes related to gamma-hydroxybutyrate (GHB) and its analogues gamma-butyrolactone (GBL) and 1,4-butanediol (1,4-BD). Drug Test Anal 2011;3(7-8):417–425.
12. Goodwin AK, et al. Behavioral effects and pharmacokinetics of gamma-hydroxybutyrate (GHB) precursors gamma-butyrolactone (GBL) and 1,4-butanediol (1,4-BD) in baboons. Psychopharmacology (Berl) 2009;204(3):465–476.
13. Verstraete AG. Detection times of drugs of abuse in blood, urine, and oral fluid. Ther Drug Monit 2004;26(2):200–205.
14. Johnston LD, et al. Monitoring the Future: National Results on Adolescent Drug Use, U.S.D.o.H.a.H. Services, Editor 2005. National Institute on Drug Abuse.
15. Hopfer C, et al. Club drug use among youths in treatment for substance abuse. Am J Addict 2006;15(1):94–99.
16. Halkitis PN, Palamar JJ. GHB use among gay and bisexual men. Addict Behav 2006;31(11):2135–2139.
17. Sumnall HR, et al. Use, function, and subjective experiences of gamma-hydroxybutyrate (GHB). Drug Alcohol Depend 2008;92(1-3):286–290.
18. Carter LP, et al. Relative abuse liability of GHB in humans: a comparison of psychomotor, subjective, and cognitive effects of supratherapeutic doses of triazolam, pentobarbital, and GHB. Neuropsychopharmacology 2006;31(11):2537–2551.
19. Zvosec DL, Smith SW. Agitation is common in gamma-hydroxybutyrate toxicity. Am J Emerg Med 2005;23(3):316–320.
20. Galloway GP, Frederick SL, Staggers F, Jr. Physical dependence on sodium oxybate. Lancet 1994;343(8888):57.
21. McDaniel CH, Miotto KA. Gamma hydroxybutyrate (GHB) and gamma butyrolactone (GBL) withdrawal: five case studies. J Psychoactive Drugs 2001;33(2):143–149.
22. Perez E, Chu J, Bania T. Seven days of gamma-hydroxybutyrate (GHB) use produces severe withdrawal. Ann Emerg Med 2006;48(2):219–220.
23. Dyer J, Roth B, Hyma B. Gamma-hydroxybutyrate withdrawal syndrome. Ann Emerg Med 2001;37:147–153.
24. Miotto K, et al. Gamma-hydroxybutyric acid: patterns of use, effects and withdrawal. Am J Addict 2001;10(3):232–241.
25. McDonough M, et al. Clinical features and management of gamma-hydroxybutyrate (GHB) withdrawal: a review. Drug Alcohol Depend 2004;75(1):3–9.
26. Okun MS, et al. GHB: an important pharmacologic and clinical update. J Pharm Pharm Sci 2001;4(2):167–175.
27. Couper FJ, Thatcher JE, Logan BK. Suspected GHB overdoses in the emergency department. J Anal Toxicol 2004;28(6):481–484.
28. van Nieuwenhuijzen PS, et al. Residual social, memory and oxytocin-related changes in rats following repeated exposure to gamma-hydroxybutyrate (GHB), 3,4-methylenedioxymethamphetamine (MDMA) or their combination. Psychopharmacology (Berl) 2010;212(4): 663–674.
29. World-Health-Organization, Pre-review of gamma-hydroxybutyric acid (GHB), 2006.
30. Kim SY, et al. High-risk behaviors and hospitalizations among gamma hydroxybutyrate (GHB) users. Am J Drug Alcohol Abuse 2007;33(3):429–438.
31. Romanelli F, Smith KM, Pomeroy C. Use of Club Drugs by HIV-Seropositive and HIV-Seronegative Gay and Bisexual Men. Top HIV Med 2003;11(1):25–32.
32. Scott-Ham M, Burton FC. Toxicological findings in cases of alleged drug-facilitated sexual assault in the United Kingdom over a 3-year period. J Clin Forensic Med 2005;12(4):175–186.
33. Stillwell ME. Drug-facilitated sexual assault involving gamma-hydroxybutyric acid. J Forensic Sci 2002;47(5): 1133–1134.
34. de Jong CA, et al. Gamma-hydroxybutyrate detoxification by titration and tapering. Eur Addict Res 2012;18(1):40–45.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


