11
CHAPTER OUTLINE
Presently, caffeine is the most widely used mood-altering drug in the world. In the United States, 87% of children and adults regularly consume foods and beverages containing caffeine (1). As a nonselective A1 and A2A adenosine receptor antagonist and mild central nervous system stimulant, caffeine produces various physiologic and psychological effects. Caffeine is not highly associated with any life-threatening illnesses, and typical daily dietary doses can be consumed under many circumstances without incident. Moreover, caffeine has valuable therapeutic effects and may offer protective effects from some diseases (e.g., Parkinson disease). However, caffeine is not completely innocuous. It has the potential to produce clinically significant negative physiologic and psychological effects (e.g., anxiety), tolerance and withdrawal, and psychiatric disorders (e.g., caffeine intoxication, caffeine use disorder). Furthermore, its coadministration with some commonly used recreational and psychotherapeutic drugs can have important clinical implications. The ubiquitous use of caffeine and its integration in daily customs and routines (e.g., coffee break) may result in a lack of appreciation for the role that caffeine plays in one’s daily subjective experiences, thus making the recognition and treatment of caffeine-associated problems particularly challenging.
Caffeine is the common name for 1,3,7-trimethylxanthine (Fig. 11-1). More than 60 types of plants containing caffeine have been identified, including coffee, tea, cola, guarana, cacao, and yerba maté. Caffeine is a member of the methyl-xanthine class of alkaloids, which includes the structurally related dimethylxanthines, theophylline, and theobromine. Caffeine in its free base form is a bitter white powder that is moderately soluble in water (21.7 mg/mL) (2). Pharmaceutical preparations of caffeine include caffeine anhydrous, caffeine sodium benzoate, and caffeine citrate.
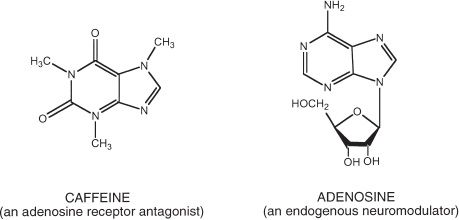
FIGURE 11-1 The chemical structure of caffeine. The chemical structure of caffeine (1,3,7-trimethylxanthine) and adenosine. Adenosine is an endogenous neuromodulator that has structural similarities to caffeine. Most of the physiologic effects of caffeine, including the central nervous system stimulant effects, are likely mediated through adenosine receptor antagonism.
Caffeine was first isolated from coffee and tea in the early 1800s, and its chemical structure was identified in 1875. Cultivation of tea in China, coffee in Ethiopia, and cacao pod in South America date back to time immemorial (3). Records of caffeine use date back at least 2,000 years for tea in China. With the development of worldwide trade in the 17th and 18th centuries, caffeinated foods and beverages rapidly spread from their original constrained geographic origins (4). In America, the protest of a British tax on tea became a symbolic focal point for revolution, resulting in the famous “Boston tea party” in 1773. After the Continental Congress passed a resolution against tea consumption, America was transformed from a predominately tea drinking land to one in which coffee was the caffeinated drink of choice (3,5). Presently, coffee is a major agricultural import of the United States, second only after oil in total value of all imports. In the late 1880s, entrepreneurs began selling carbonated beverages with caffeine, and the last century has seen a steady increase in caffeinated soft drink consumption. One of the first energy drinks, Red Bull, was introduced in Austria in 1987 and in the United States in 1997. Presently, energy drinks represent a multibillion dollar market with hundreds of different brands available to consumers (6). Liquid dietary supplements with very high concentrations of caffeine (e.g., 280 mg/2 oz) are also being mass marketed in the United States and elsewhere. Some countries ban the sale of energy drinks or strictly regulate their caffeine content and/or labeling because of concerns about negative health effects. However, the regulation of energy drinks in the United States has been heretofore quite lax (6). The increasing incidence of adverse events after consumption of energy drinks (7) and recent claims that energy drinks have contributed to sudden deaths in young people have led to public scrutiny and an ongoing FDA investigation on the safety of energy drinks (8).
The most recent large-scale published epidemiologic data on caffeine consumption in the United States was collected nearly 15 years ago. Based on the Continuing Survey of Food Intakes by Individuals in 1994–1996 and 1998, it is estimated that 87% of the population in the United States 2 years and older regularly consume caffeine with an average daily consumption of about 193 mg (1). The highest consumption is among individuals aged 35 to 64 years (1). Mean daily intake of caffeine for adult caffeine consumers has been estimated to be about 280 mg in the United States, with higher intakes estimated for consumers in the United Kingdom and Denmark (9). In the United States, coffee and soft drinks are the major dietary sources of caffeine while energy drink consumption continues to sharply increase each year. More than half of the adult U.S. population consumes coffee every day. A recent survey of active duty U.S. Army soldiers found that 82% were regular caffeine consumers, consuming on average 347 mg/d. Coffee was the most common source of caffeine in general; however, younger soldiers consumed most of their caffeine from energy drinks (10). A 2010 study by the Centers for Disease Control involving a random sample of male Marine and Army platoons in Afghanistan reported that 45% consumed energy drinks on a daily basis with 14% consuming three or more daily (11).
There have been no large-scale epidemiologic surveys on caffeine-related psychiatric disorders. One random-digit telephone survey in Vermont found that 7% of current caffeine users had symptoms consistent with DSM-IV caffeine intoxication in the past year (12). Prior studies that have used ambiguous criteria and have focused on special populations (e.g., psychiatric patients, college students) have reported caffeine intoxication rates ranging from 2% to 19%. A large-scale twin study found that 29% reported having felt ill or shaky or jittery after consuming caffeinated beverages (13).
The Vermont-based random-digit telephone survey also assessed the incidence of caffeine withdrawal using DSM-IV research criteria (12). In that study, 44% of caffeine users reported having stopped or reduced caffeine use for at least 24 hours in the past year. Of those, 41% reported that they experienced one or more DSM-IV–defined caffeine withdrawal symptoms. Among individuals who attempted to permanently stop using caffeine, at least 71% reported experiencing DSM-IV–defined withdrawal symptoms, and 24% met criteria by reporting headache plus other symptoms that interfered with functioning.
This same study examined the prevalence of substance dependence applied to caffeine in the general population and found that 30% of caffeine consumers fulfilled DSM-IV diagnostic criteria in the past year (12). The most commonly endorsed symptom was desire or unsuccessful efforts to cut down or control use (56%).
As shown in Table 11-1, sources of caffeine include beverages, foods, dietary supplements, and over-the-counter and prescription medications. Estimating caffeine exposure can be challenging because of the wide variety of products that contain caffeine, large differences in common serving sizes, and great variability in caffeine content across products of the same type. For example, a 12-oz cup of coffee may contain anywhere from 107 to 420 mg of caffeine. The U.S. Food and Drug Administration (FDA) limits the amount of caffeine contained in soft drinks to 0.02%, or 71.5 mg/12 oz. However, at this time, such limits do not apply to energy drinks, which can vary more than 10-fold in caffeine content across brands (e.g., 36 to 375 mg per can or bottle). In recent years, there has been widespread marketing of highly caffeinated dietary supplements (e.g., 5-Hour Energy), and manufacturers have begun to add caffeine to foods that have traditionally not contained caffeine (e.g., potato chips, jelly beans). Caffeinated beer, malt beverages, and hard liquors were available to consumers for a short while before the FDA banned the sale of these products in 2010 due to safety concerns (14). However, it is an increasingly common practice, especially among young people, to mix energy drinks with alcohol (e.g., Red Bull and vodka) (14).
TABLE 11-1 CAFFEINE CONTENT OF COMMON FOODS AND MEDICATIONS
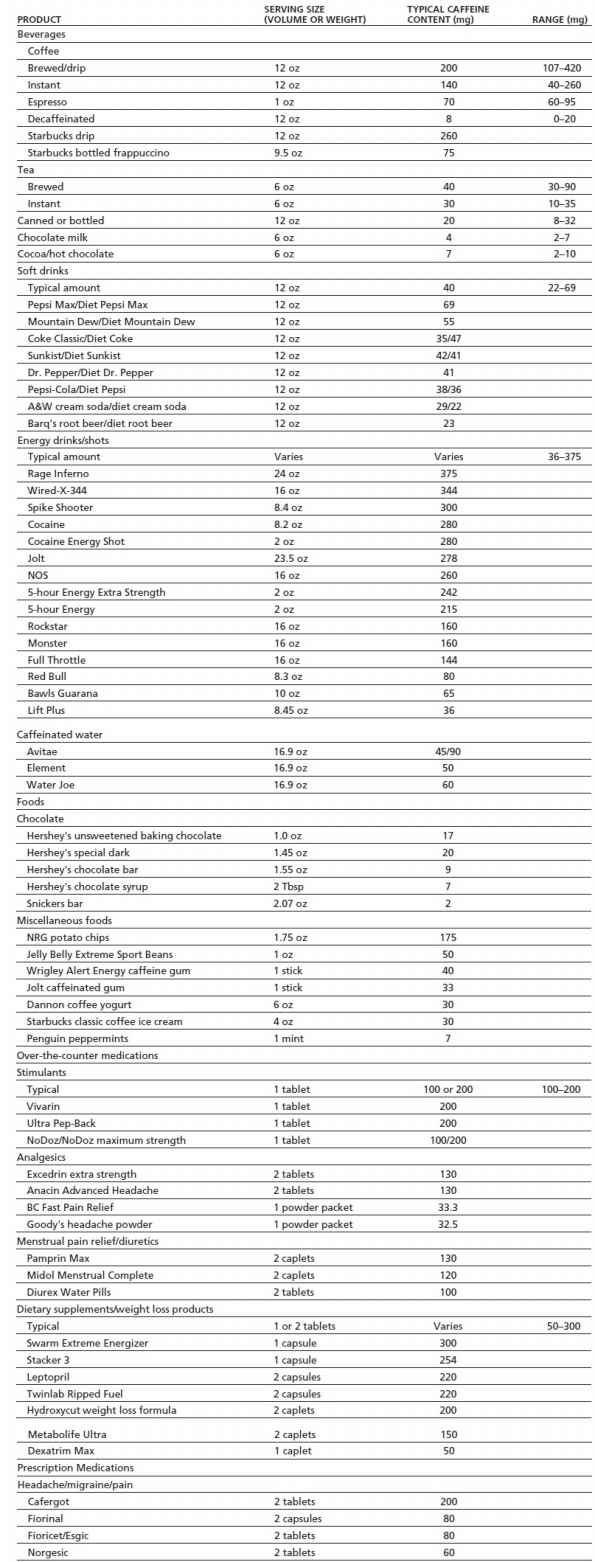
Caffeine values for all brand name products were obtained directly from product labels, the manufacturer’s Web site, or customer service department with the exception of 5-hour Energy that was obtained from Consumer Reports Magazine, December 2012.
Data from Barone JJ, Roberts HR. Caffeine consumption. Food Chem Toxicol 1996;34:119–129; McCusker RR, Fuehrlein B, Goldberger BA, et al. Caffeine content of decaffeinated coffee. J Anal Toxicol 2006;30:611–613; and McCusker RR, Goldberger BA, Cone EJ. Caffeine content of specialty coffees. J Anal Toxicol 2003;27:520–522.
As a mild central nervous system stimulant, caffeine is widely used to increase energy and prevent sleepiness. Because of its analgesic-enhancing effects, caffeine is found in the formulations of a wide range of over-the-counter and prescription analgesic products that are frequently used to treat various types of pain, including migraine (15). A recent Cochrane Review concluded that the addition of caffeine to pain medication increased the percentage of people having a significant analgesic effect by 5% to 10% (16). Not surprisingly, caffeine alone is the most effective treatment for caffeine withdrawal headache, with prophylactic caffeine administration recognized as an effective way to prevent postsurgical headaches in caffeine consumers (17,18). As a respiratory stimulant, caffeine is used to treat apnea in neonates and infants (19). Intravenous caffeine has also been administered before electroconvulsive therapy to lengthen seizure duration; however, its therapeutic merits are unclear (20). Caffeine has been used to treat postprandial hypotension, although its effectiveness has been questioned (21). Because of its lipolytic and thermogenic effects, caffeine is commonly used in weight loss preparations and nutritional supplements. Caffeine is also used to facilitate athletic performance because of its ergogenic effects, although meaningful enhancements may be limited to well-conditioned athletes (22).
The primary cellular site of action of caffeine is the adenosine receptor. Adenosine is an endogenous purine nucleoside found throughout the brain. It is formed from the breakdown of adenosine triphosphate (ATP) and modulates a variety of central and peripheral nervous system effects. Caffeine is structurally similar to adenosine (see Fig. 11-1). As a competitive A1 and A2A adenosine receptor antagonist, caffeine produces a variety of effects that are opposite to the effects of adenosine (e.g., central nervous stimulation, vasoconstriction).
More specifically, caffeine produces its motor and reinforcing effects by releasing the pre- and postsynaptic brakes that adenosine imposes on striatal dopaminergic neuro-transmission. At the presynaptic level, caffeine induces dopamine release by a glutamate-independent mechanism, by targeting adenosine A1 receptors localized in striatal dopaminergic terminals. Furthermore, caffeine induces dopamine release by a glutamate-dependent mechanism, by targeting A1 receptors that form heteromers with adenosine A2A receptors in striatal glutamatergic terminals. These pre-synaptic effects of caffeine are potentiated by postsynaptic effects, which depend on the existence of strong antagonistic adenosine–dopamine receptor–receptor interactions. These interactions depend on the ability of A1 and A2A receptors to form heteromers with dopamine D1 and D2 receptors, respectively. The effects of caffeine on sleep do not seem to be dopamine dependent and are related to the ability of adenosine to act as an endogenous sleep-promoting substance by acting on not only A1 and A2A receptors localized in the brainstem, basal forebrain, and hypothalamic areas but also A2A receptors localized in the ventral part of the striatum. These effects are discussed in greater detail in the following sections.
Adenosine A1 and A2A Receptor Antagonism
Among the four cloned adenosine receptors (A1, A2A, A2B, and A3 receptors), A1 and A2A receptors are the ones predominantly expressed in the brain. Caffeine is a nonselective adenosine receptor antagonist, with similar in vitro affinities for A1, A2A, and A2B receptors and with much lower affinity for A3 receptors (23,24). A1 and A2A receptors are the preferential targets for caffeine in the brain, because physiologic extracellular levels of adenosine are sufficient to occupy and, therefore, stimulate A1 and A2A receptors. On the other hand, A2B receptors have a lower affinity for adenosine, and they are only activated by high pathologic extracellular levels of adenosine (23). A1 receptors are widely expressed in the brain, including the striatum, whereas A2A receptors are highly concentrated in the striatum (23,25). The striatal localization of both receptors seems to underlie the motor-activating and reinforcing effects of caffeine (See Postsynaptic Mechanisms: Adenosine–Dopamine Receptor Interactions). On the other hand, A1 receptors localized in the brainstem and basal forebrain and A2A receptors localized in the hypothalamus have been suggested to be involved in caffeine effects on sleep regulation.
There has been a long debate about the preferential involvement of A1 and A2A receptors in the psychostimulant effects of caffeine (26,27). By comparing both quantitative and qualitative aspects of the motor activity induced by caffeine and selective A1 and A2A receptor antagonists, recent studies have clearly shown that caffeine, when administered acutely, shows a profile of a nonselective adenosine receptor antagonist with preferential A1 receptor antagonism (28,29). Also, recent drug discrimination experiments support a key role of A1 receptors in the psychostimulant effects of acutely administered caffeine (24). Importantly, chronic exposure to caffeine differentially modifies its motor effects dependent on A1 and A2A receptor blockade. Thus, chronic exposure to caffeine in the drinking water of rats results in partial tolerance to the motor effects of an additional acute administration of caffeine and total cross-tolerance to the motor effects of an A1 but not an A2A receptor antagonist (29). This indicates that tolerance to the effects of A1 receptor blockade is mostly responsible for the tolerance to the motor-activating effects of caffeine and that the residual motor-activating effects of caffeine in tolerant individuals might be mostly because of A2A receptor blockade.
An important amount of experimental evidence supports a key role of dopamine in the psychostimulant effects of caffeine in animals and humans. For instance, dopamine depletion or blockade of dopamine receptors significantly impairs the motor and discriminative stimulus effects of caffeine (30,31). Most probably, the same basic dopamine-mediated mechanisms are involved in the motor-activating and reinforcing effects of caffeine, as happens with classical psychostimulants (32,33). The key to understanding these mechanisms of action is to appreciate how adenosine modulates dopaminergic neurotransmission in the brain.
The effects of caffeine on sleep regulation do not seem to be dopamine dependent, and they are related to its ability to antagonize the sleep-promoting effects of adenosine (34–37). Although previous results suggested that A1 receptors are involved in sleep regulation by inhibiting ascending cholinergic neurons of the basal forebrain (34,37), more recent studies, which include experiments with global and selective genetic blockade of A2A receptors in mice, indicate that A2A receptors play a crucial role in the sleep-promoting effects of adenosine and the arousal-enhancing effects of caffeine (35,36,38). Some of these A2A receptors are probably localized in the ventrolateral preoptic area of the hypothalamus, and their stimulation promotes sleep by inducing gamma-aminobutyric acid (GABA) release in the histaminergic tuberomammillary nucleus, thereby inhibiting the histaminergic arousal system (35,36). Furthermore, A2A receptors localized in the ventral part of the striatum (shell of the nucleus accumbens) also seem to be involved (38). This striatal area directly and indirectly connects with hypothalamic and mesencephalic nuclei of origin of several ascending arousal systems (38).
Adenosine is a key modulator of dopaminergic and glutamatergic neurotransmission in the striatum (39). Recent studies suggest that astroglia plays a fundamental role in the formation of extracellular adenosine, which can influence synaptic transmission. Astrocytes express glutamate receptors (mostly metabotropic) and ATP receptors that, when activated, induce astrocytes to release glutamate and ATP (40,41). Astroglial-released ATP is then converted to adenosine in the extracellular space by means of ectonucleotidases (42). Finally, there is an increasing amount of data suggesting the existence of a neurotransmitter-like formation of adenosine, a synaptic pool of adenosine. Adenosine can be produced from ATP coreleased with glutamate, which is also metabolized to adenosine by means of ectonucleotidases (25,39). GABAergic enkephalinergic and dynorphinergic medium-sized spiny neurons constitute more than 90% of the striatal neuronal population (43). In the striatum, A2A receptors are localized postsynaptically in the dendritic spines of enkephalinergic medium-sized spiny neurons, colocalized with D2 receptors and presynaptically in glutamatergic terminals, colocalized with A1 receptors (25,39,44,45). In addition to the glutamatergic terminals, A1 receptors are localized in a fraction of dopaminergic nerve terminals (46) and, postsynaptically, in the dendritic spines of dynorphinergic medium spiny neurons, colocalized with D1 receptors (45). Antagonistic interactions between A2A and D2 receptors modulate the function of the enkephalinergic medium spiny neuron, and antagonistic interactions between A1 and D1 receptors modulate the function of the dynorphinergic medium spiny neuron (47,48). This gives the explanation at the neuronal level of an important number of pharmacologic findings indicating a selective modulation of A1 and A2A receptor ligands on D1 and D2 receptor–mediated behavioral effects, respectively (30,45,49).
Postsynaptic Mechanisms: Adenosine–Dopamine Receptor Interactions
The molecular mechanisms responsible for the selective antagonistic A1–D1 and A2A–D2 receptor interactions have been found to involve allosteric interactions in A1–D1 and A2A–D2 receptor heteromers (44,50). Thus, there is compelling evidence for the existence of A1–D1 and A2A–D2 receptor heteromers in artificial cell systems and in the striatum (44,50). In the A2A–D2 heteromer, the stimulation of the A2A receptor decreases the binding of dopamine to the D2 receptor (51). This intramembrane interaction controls neuronal excitability and, consequently, neuronal firing and neurotransmitter (GABA) release by the GABAergic enkephalinergic neuron (25,48,52,53). Furthermore, there is a reciprocal antagonistic A2A–D2 receptor interaction by which D2 receptor stimulation counteracts the effects induced by A2A receptor stimulation at the level of adenylyl cyclase and, consequently, at the level of protein phosphorylation and gene expression (54–56).
The allosteric interaction in the A2A–D2 heteromer seems to play a key role in the motor-activating effects of caffeine. In fact, this A2A–D2 receptor interaction has been given a lot of attention in the literature, more recently with the application of A2A receptor antagonists as an adjuvant therapy for levodopa in Parkinson disease (57). But, as mentioned previously, A1–D1 receptor interactions are also of significant functional and pharmacologic importance. Thus, A1 receptor antagonists selectively potentiate the motor- activating effects of D1 receptor–mediated motor activation in different animal models (47,58). Similar to what happens with the A2A–D2 receptor heteromers, A1 and D1 receptors antagonistically interact both allosterically and at the adenylyl cyclase level. However, in this case, the interactions are not reciprocal, and stimulation of A1 receptors inhibits both the binding of dopamine to the D1 receptor (59,60) and the D1 receptor–mediated activation of cAMP–PKA signaling pathway and the expression of genes, such as c-fos and preprodynorphin (60,61).
Presynaptic Mechanisms: Modulation of Glutamate and Dopamine Release
Studies have repeatedly shown that A1 and A2A receptors exert opposite modulatory roles on extracellular levels of glutamate and dopamine in the striatum, with activation of A1 receptors inhibiting and activation of A2A receptors stimulating glutamate and dopamine release (26,27). More recently, it has been shown that systemic or striatal administration of caffeine or an A1, but not an A2A, receptor antagonist produces a significant increase in the extracellular concentrations of glutamate and dopamine in the ventral striatum, particularly in the most medial part, the medial shell of the nucleus accumbens (62,63). It was hypothesized that dopamine release mostly depended on glutamate release induced by blockade of A1 receptors localized in glutamatergic terminals and on stimulation of ionotropic glutamate receptors localized in dopaminergic terminals (63,64). Importantly, chronic administration of caffeine in the rat’s drinking water completely counteracted the effects of caffeine or an A1 receptor antagonist on dopamine and glutamate, whereas the effect of an A2A receptor antagonist was not modified (62). Thus, these biochemical changes are consistent with the studies on motor activity described previously (29), strongly suggesting an involvement of presynaptic mechanisms in the psychostimulant effects of caffeine. The ability of caffeine to release dopamine in the nucleus accumbens was questioned by one research group (65), but a recent study demonstrated the existence of sub-regional differences in the effect of A1 receptor blockade in different parts of the nucleus accumbens and other striatal areas, most probably related to subregional differences in the level of tonic activation by endogenous adenosine (46). Furthermore, glutamate-independent mechanisms were also found to be involved in A1 receptor blockade–mediated striatal dopamine release, which depended on A1 receptors localized in dopaminergic nerve terminals (46).
Dopamine release in the very medial striatal compartments seems to be involved in both motor-activating and reinforcing effects of psychostimulants (32). Therefore, the motor and reinforcing effects of caffeine most probably depend on the pre- and postsynaptic dopaminergic mechanisms mentioned previously (striatal dopamine release and adenosine–dopamine receptor–receptor interactions) taking place in the medial striatal compartments. At least two factors related to the dopamine-releasing effects of caffeine could explain its weaker reinforcing effects as compared with other psychostimulants: its specific subregional effects (46) and the development of tolerance (62).
Under chronic caffeine exposure, different factors might contribute to the predominant tolerance of the effects of A1 receptor blockade. Up-regulation of A1 receptors has been repeatedly demonstrated (29,66–68), but its significance as a mechanism involved in caffeine tolerance has been seriously questioned (69). Modifications in the function of A1–A2A receptor heteromers might play a key role in the development of caffeine tolerance. Thus, radioligand- binding experiments have shown that chronic treatment with caffeine causes an increase in the potency of A2A receptor agonist–mediated inhibition of A1 receptor agonist binding and a significant reduction in the affinity of the striatal A2A receptor for caffeine (70). An additional factor that might play a significant role in caffeine tolerance is the significant increase in plasma and extracellular concentrations of adenosine with chronic caffeine exposure (71). The higher adenosine levels and lower affinity of the A2A receptor for caffeine could allow endogenous adenosine to stimulate A2A receptors even in the presence of caffeine, which would not reach enough concentration to compete with adenosine for binding A2A receptors. Under these conditions, A1 receptor signaling in the A1–A2A receptor heteromer would be expected to be chronically turned down, because of its continuous blockade by caffeine and because of the strong A2A receptor–mediated inhibition of A1 receptor agonist binding. On the other hand, an additional administration of caffeine would be expected to produce a blockade of the residual A2A receptor signaling.
Absorption and Distribution
Caffeine is rapidly and completely absorbed after oral administration, with peak levels reached in 30 to 45 minutes (72). It is readily distributed throughout the body, with concentrations in blood correlating with those in saliva, breast milk, amniotic fluid, fetal tissue, semen, and the brain (73). Binding to plasma proteins is estimated to range between 10% and 35% (74). Saliva caffeine concentrations, which often exceed 75% of plasma concentrations, are used as a noninvasive alternative to serum monitoring.
Metabolism
Caffeine metabolism is complex, with more than 25 metabolites identified in humans (75). Caffeine is metabolized by the cytochrome P-450 liver enzyme system. In particular, the CYP1A2 isoenzyme demethylates caffeine to three biologically active dimethylxanthines: paraxanthine, theobromine, and theophylline, accounting for 80%, 10%, and 4% of caffeine metabolism, respectively (74). The large amounts of paraxanthine, coupled with the demonstration of similar sympathomimetic effects of paraxanthine and caffeine, suggest that paraxanthine needs to be considered in understanding the clinical pharmacology of caffeine (76).
Elimination
On average, caffeine half-life is 4 to 6 hours. However, there are wide individual differences in rates of caffeine elimination (74), which are due in large part to CPY1A2 genetic variation across individuals (77). An important implication of the central role of cytochrome P-450 liver enzymes in metabolizing caffeine is that drugs or conditions that affect this metabolic system significantly alter caffeine elimination. Caffeine’s half-life is prolonged with liver disease (74), presumably because of lower CYP1A2 activity (78). Although caffeine half-life does not differ between younger and older adults (79), caffeine half-life is markedly increased in premature and full-term newborns (half-life of 80 to 100 hours) whose liver enzyme capacity is not completely developed until about 6 months of age (80). Cigarette smoking, which induces liver enzymes, decreases caffeine half-life by as much as 50% (81). Numerous compounds have been shown to inhibit caffeine metabolism including oral contraceptives, cimetidine, some quinoline antibiotics (e.g., ciprofloxacin), fluvoxamine, and mexiletine (75,82). Caffeine half-life increases markedly during the end of pregnancy (83), which could increase the risk of caffeine toxicity among women who maintain high levels of caffeine use during pregnancy (84).
At moderate dietary dose levels, caffeine increases systolic and diastolic blood pressure (85). Caffeine tends to have no effect (86) or to reduce heart rate in humans (87). Caffeine constricts blood vessels in the head and neck. Caffeine stimulates gastric acid secretions and is a colonic stimulant, with caffeinated coffee producing colonic motor activity similar to that produced by a meal (88). It is a diuretic, increasing urine volume 30% or more for several hours after ingestion (89). Caffeine also increases detrusor pressure on the bladder of patients with complaints of urinary urgency and confirmed detrusor instability (90). Caffeine is a respiratory stimulant (91) and a bronchodilator at high doses (92). Caffeine is ergogenic across a variety of exercise situations, and in particular during prolonged exercise (93), with activity potentially mediated via multiple mechanisms, including effects on muscle contractility (94). Caffeine increases plasma epinephrine, norepinephrine, renin, and free fatty acids, particularly in nontolerant individuals (76,95,96). It also increases adrenocorticotropic hormone and cortisol (97–99). Caffeine increases insulin levels in healthy subjects (100) and increases postprandial glucose and insulin responses among patients with type 2 diabetes who are habitual coffee drinkers (100,101).
Human laboratory studies demonstrate that single low to moderate doses of caffeine typically produce a profile of positive subjective effects, including increased well-being, happiness, energy, arousal, alertness, and sociability (102). Positive effects are more likely to be observed in studies that require participants abstain from caffeine before testing and that administer a caffeine dose in the range of typical dietary consumption (e.g., 20 to 200 mg) (103). Physical dependence also increases the positive mood effects of caffeine, likely through suppression of low-grade withdrawal symptoms (104,105). However, positive mood effects occur among nonhabitual users and those maintained on a caffeine-free diet, as well as under conditions of minimal caffeine deprivation (106–111).
Negative subjective effects of caffeine are more likely to be observed after acute doses of caffeine greater than 200 mg and include anxiety, nervousness, jitteriness, negative mood, upset stomach, sleeplessness, and “bad effects” (102). Individual differences in use, sensitivity, and tolerance seem to play an important role in the likelihood and severity of negative subjective effects (112). Acute doses of caffeine greater than 200 mg can increase anxiety ratings in both individuals with anxiety disorders (113–115) and nonclinical samples (116–118), with greater effects among the former (113–115,119,120). Higher doses of caffeine can also elicit panic attacks (116,121,122). The DSM-5 (Diagnostic and statistical manual of mental disorders, 5th ed.) recognizes Caffeine-Induced Anxiety Disorder, which is defined as anxiety symptoms or an anxiety disorder (e.g., generalized anxiety disorder) caused by caffeine use (123).
Caffeine can also have negative effects on planned sleep. Caffeine delays sleep onset, reduces total sleep time, alters the normal stages of sleep, and decreases the reported quality of sleep (124–126). Even caffeine taken early in the day can disrupt nighttime sleep (127). Caffeine-induced sleep disorder is a diagnosis recognized by DSM-5 characterized by a prominent sleep disturbance etiologically related to caffeine use (123).
A large number of studies have examined the effects of caffeine on human performance. The most consistent generality to emerge is that caffeine reliably increases performance on task performance that has been degraded by fatigue (e.g., under conditions of sleep deprivation or prolonged vigilance) (73,128,129). Although the results are variable (73,130), and observed effects are often small (130), compared with placebo, caffeine at normal dietary doses may improve tapping speed, reaction time, sustained attention (or vigilance), and perhaps focused attention (130,131). The effect of caffeine on various memory tasks has also been investigated but is inconclusive (130). A growing literature on the effects of caffeine on exercise performance suggests that, relative to placebo, caffeine can enhance performance during long-term (30 to 60 minutes) aerobic exercise (22,93), can reduce ratings of perceived exhaustion (132,133), and can improve speed or power output in simulated race conditions (22,134). Some studies have also demonstrated caffeine-induced enhancement during short-term, high-intensity exercise (134,135), but these effects are generally more difficult to demonstrate (22,133,136). A problem in interpreting the effects of caffeine on performance is that most studies have compared the effects of caffeine and placebo on the performance of habitual caffeine users who have been required to abstain from caffeine, usually overnight. Thus, improvements in performance after caffeine relative to placebo may simply reflect a relief of withdrawal symptoms or restoration to baseline performance (106,137). However, some studies have shown caffeine-related performance enhancements among light nondependent caffeine consumers and non-consumers (107), nonwithdrawn caffeine consumers (138) as well as caffeine consumers after a protracted period of abstinence (139). Based on the preclinical literature, which clearly documents the behavioral stimulant effects of caffeine, it seems quite likely that caffeine enhances human performance on some types of tasks (e.g., vigilance), especially among nontolerant individuals. Among high-dose habitual caffeine consumers, performance enhancements above and beyond withdrawal reversal effects are perhaps modest at best (104).
DISCRIMINATIVE STIMULUS EFFECTS
Carefully controlled double-blind studies show that the majority of individuals (>80%) can learn to reliably discriminate caffeine from placebo (140), using initial caffeine doses ranging from 100 to 320 mg. Studies that have explicitly trained discrimination of progressively lower caffeine doses show that caffeine doses as low as 1.8 to 10 mg can be reliably discriminated by some participants (109,110,141). These studies documenting that caffeine can produce reliable discriminative effects at very low doses are consistent with studies showing that low doses of caffeine (e.g., 9 to 12.5 mg) can produce improvements in behavioral performance (142,143). Drug discrimination studies in humans and animals have demonstrated both similarities and differences between caffeine and other stimulant drugs (e.g., cocaine, d-amphetamine, methylphenidate) (102).
The circumstantial evidence for caffeine functioning as a reinforcer is compelling. Caffeine is the most widely self-administered mood-altering drug in the world and, historically, repeated efforts to restrict or eliminate consumption of caffeinated foods have been completely unsuccessful (3,5). Several carefully controlled research studies over the past 20 years provide unequivocal evidence for the reinforcing effects of caffeine (102). Caffeine reinforcement has been demonstrated with various participant populations, using a variety of methodologic approaches (e.g., choice procedures, ad libitum self-administration), and across different caffeine vehicles (e.g., coffee, soft drinks, capsules). The average incidence of caffeine reinforcement across studies in normal caffeine users is approximately 40%, with higher rates observed (i.e., 82% to 100%) among certain subsamples such as high caffeine consumers, those with histories of drug or alcohol abuse, in studies involving repeated exposure to caffeine and placebo test conditions before reinforcement testing (102,144), and in the context of having to perform a vigilance task after drug administration (111). Doses as low as 25 mg per cup of coffee and 33 mg per serving of soft drink function as reinforcers (145–147). Doses greater than 50 or 100 mg tend to decrease choice or self-administration, with relatively high doses of caffeine (e.g., 400 or 600 mg) sometimes producing significant caffeine avoidance (148). The subjective effects of caffeine covary with caffeine reinforcement (103). Positive subjective effects of caffeine predict the subsequent choice of caffeine relative to placebo, and negative subjective effects predict the subsequent choice of placebo relative to caffeine (149). Furthermore, caffeine users who report negative effects of placebo (i.e., withdrawal symptoms) also tend to choose caffeine over placebo (150). Avoidance of caffeine withdrawal symptoms clearly plays a central role in the reinforcing effects of caffeine among regular caffeine consumers. For example, in studies that prospectively manipulated caffeine physical dependence, subjects chose caffeine more than twice as often when they were physically dependent than when they were not physically dependent (151,152).
A series of studies has used a conditioned flavor preference paradigm to provide suggestive evidence of caffeine reinforcement. These studies have shown that abstinent caffeine consumers who are repeatedly given a novel flavored drink paired with either caffeine (e.g., 70 mg) or placebo, rate the caffeine-paired drink as more pleasant or preferred, and the placebo-paired drink as less so (153,154). Caffeine paired with a novel-flavored yogurt increased preference for the yogurt suggesting that food preferences may be influenced by caffeine coadministration (155). The ability of caffeine to produce changes in flavor liking also appears to be driven by the alleviation of withdrawal symptoms among habitual caffeine consumers (i.e., negative reinforcement) (156). In the natural environment, the development of such conditioned flavor preferences over many days of self-administration likely play an important role in engendering strong consumer preferences for specific types and brands of caffeine-containing beverages.
Caffeine reinforcement has also been investigated in animals using self-injection or conditioned place preference paradigms. Animals will self-inject caffeine, sometimes showing patterns of irregular intake (e.g., high rates of intake alternating irregularly with periods of low intake) (102). Somewhat consistent with self-injection studies of nicotine, caffeine does not reliably maintain self-administration across animals and studies to the extent that is observed in studies of classic abused stimulants (e.g., cocaine, d-amphetamine). Low doses of caffeine produce conditioned place preference in rats, whereas higher doses produce clear place avoidance (102). Similar to the self-injection data, a direct comparison of place preference between caffeine and cocaine suggested that cocaine has greater reinforcing effects (157). A recent study found that caffeine pretreatment in rats increased the reinforcing value of nondrug stimuli (i.e., sucrose, visual stimulus) but that tolerance developed to this effect in the 15 days of testing (158).
The degree of tolerance development to caffeine can be expected to depend on the caffeine dose, the dose frequency, the number of doses, and the individual’s elimination rate (159). Tolerance has been clearly demonstrated in both animals and humans; however, quantitative parametric information is quite fragmentary. Complete tolerance does not occur at low daily dietary doses. Very high doses of caffeine (750 to 1,200 mg/d spread throughout the day), administered daily, produce “complete” tolerance (i.e., caffeine effects are no longer different from baseline or placebo) to some but not all effects. For example, in one study, differences in ratings of mood and other subjective effects were observed between participants given 300 mg of caffeine three times daily and those given placebo three times daily only during the first 4 days of an 18-day study, clearly suggesting tolerance development (150). One within-subjects study administered 400 mg caffeine or placebo daily for 21 days and found that tolerance developed to the subjective effects of caffeine but not its effects on cerebral blood flow and EEG (160). Tolerance develops to the sleep- disrupting effects of caffeine (161,162) and to physiologic effects including diuresis, parotid gland salivation, increased metabolic rate (oxygen consumption), increased plasma nor-epinephrine and epinephrine, and increased plasma renin activity (140). Tolerance also develops to blood pressure increases caused by caffeine ingestion; however, controlled research has shown that tolerance to such pressor effects is incomplete (163–165).
Studies in animals have demonstrated complete, insurmountable tolerance to high doses of caffeine. Caffeine tolerance occurs across different animal species (e.g., mice, rats, monkeys) and a wide range of experimental measures (e.g., locomotor activity, schedule-controlled responding, reinforcement thresholds for electrical brain stimulation, caffeine-induced seizure thresholds, discriminative responding in caffeine-trained animals).
Reports of caffeine intoxication can be found in the medical literature dating back to the 1800s (166). “Caffeinism” is an older term that has been used to describe negative or toxic consequences of caffeine resulting from acute or chronic use (167). Unlike many other drugs of dependence, but similar to nicotine, the high-dose intoxicating effects of caffeine are not usually sought out by users.
Caffeine intoxication is a diagnosis in DSM-5 (123) and in the ICD-10 (International Statistical Classification of Diseases and Related Health Problems) (168). Caffeine intoxication is defined by the DSM-5 as the emergence of five or more of the following symptoms after excess ingestion of caffeine (>250 mg): restlessness, nervousness, excitement, insomnia, flushed face, diuresis, gastrointestinal disturbance, muscle twitching, rambling flow of thought and speech, tachycardia or cardiac arrhythmia, inexhaustibility, and psychomotor agitation. In addition, there have been reports of patients with caffeine intoxication having fever, irritability, sensory disturbances, tachypnea, and headaches (166). Individual sensitivity (e.g., metabolic differences) and tolerance likely influence the dose effects.
Symptoms of caffeine intoxication overlap with other medical and psychiatric disorders, and thus, identifying recent ingestion of caffeine is critical in identifying caffeine intoxication. Serum or saliva assays of caffeine can be used to confirm caffeine use. Caffeine intoxication should be considered as a differential diagnosis when assessing possible intoxication and withdrawal from other drugs, medication side effects (e.g., akathisia), other psychiatric disorders (e.g., anxiety disorders, mania, sleep disturbances), and somatic disorders (e.g., arrhythmia, hyperthyroidism). Caffeine intoxication resolves rapidly (consistent with caffeine’s half-life of 4 to 6 hours) and usually with no long-lasting consequences. However, medical treatment and monitoring is necessary when significant caffeine overdose occurs. Caffeine can be lethal after ingestion of very high doses (i.e., about 5 to 10 g), and there is documentation of accidental overdose and suicide by caffeine ingestion, usually in the form of pills (102). It has been suggested that the emergence of highly caffeinated energy drinks in recent years may be increasing the incidence of caffeine intoxication. For example, a series of case reports based on California poison control data found that a particular energy drink containing 250 mg of caffeine was repeatedly implicated in caffeine intoxication reports, mostly among young people. The most common symptoms reported were hypertension, tachycardia, jitteriness, agitation, tremors, nausea, vomiting, and dizziness (169). A recent report by the Drug Abuse Warning Network found that the number of emergency department (ED) visits involving energy drinks doubled from 2007 to 2011 with most visits involving males and individuals between the ages of 18 and 25. Of the 20,783 energy drink–related ED visits in 2011, 58% involved only energy drinks and 42% involved energy drinks combined with other drugs or alcohol (13%) (7). Furthermore, at this time, the FDA is investigating a number of unexpected deaths among young people after consumption of energy drinks that some have attributed to high doses of caffeine.
Reports of caffeine withdrawal in the medical literature date back more than 175 years, and presently, the caffeine withdrawal syndrome is well characterized. A 2004 comprehensive review of carefully controlled caffeine withdrawal research identified 13 symptoms with a strong empirical basis (Table 11-2). The symptoms were conceptually grouped into the following five categories and later validated by a factor analysis of symptoms reported by habitual caffeine consumers undergoing a trial of acute caffeine abstinence (170): (a) headache; (b) fatigue or drowsiness; (c) dysphoric mood, depressed mood, or irritability; (d) difficulty concentrating; and (e) flu-like somatic symptoms nausea, vomiting, and muscle pain/stiffness. Caffeine withdrawal syndrome is currently defined by the DSM-5 as the presence of three or more of these five signs or symptoms within 24 hours after abrupt cessation of or reduction in caffeine use. Symptoms must cause clinically significant distress or impairment in social, occupational, or other important areas of functioning. A review of research found that habitual caffeine consumers undergoing acute caffeine abstinence experience clinically significant distress and/or functional impairment about 13% of the time. Reports of functional impairment include but are not limited to being unable to care for children or go to work, school, or church (171).
TABLE 11-2 EMPIRICALLY VALIDATED SIGNS AND SYMPTOMS RESULTING FROM CAFFEINE ABSTINENCE
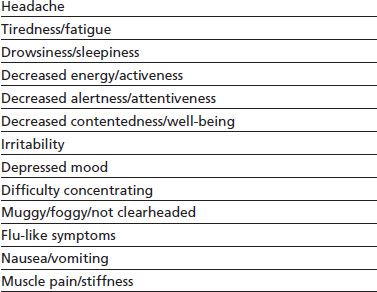
From Juliano LM, Griffiths RR. A critical review of caffeine withdrawal: empirical validation of symptoms and signs, incidence, severity, and associated features. Psychopharmacology (Berl) 2004;176:1–29.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


