7
CHAPTER OUTLINE
■ SUBSTANCES INCLUDED IN THIS CLASS
■ FORMULATIONS AND METHODS OF USE
■ NEUROBIOLOGY (MECHANISMS OF ADDICTION)
Alcohol is a chemical name for a group of related compounds that contain a hydroxyl group (-OH) bound to a carbon atom. The form of alcohol that is voluntarily consumed by humans is ethyl alcohol or ethanol and consists of two carbons and a single hydroxyl group (written as C2H5OH or C2H6O). Unless otherwise noted, the term alcohol is used throughout this chapter to mean ethanol.
SUBSTANCES INCLUDED IN THIS CLASS
In terms of human consumption, all commercially available alcoholic beverages contain ethyl alcohol with concentrations depending upon the type of beverage. Beverages made by fermentation of sugar-containing fruits and grains include beer (3% to 8% ethanol by volume) and wines (11% to 13% ethanol by volume). Spirits are produced after distillation and generally contain at least 30% ethanol. Ethanol can be concentrated by simple distillation up to approximately 95%, while pure ethanol requires addition of benzene or related substances or desiccation using glycerol. Denatured alcohol contains additives or toxins to prevent human consumption. Rubbing alcohol is prepared from denatured alcohol or isopropyl alcohol and is used for topical purposes.
FORMULATIONS AND METHODS OF USE
A bewildering array of alcoholic beverages is available for consumption, and these products contain a wide range of alcohol concentrations. In the United States, a standard alcoholic drink is defined as one that contains 0.6 fluid ounces of alcohol. Thus, this amount of alcohol is typically contained in 12 oz of beer, 5 oz of wine, or 1.5 oz of distilled spirits (40% ethanol by volume) although this can vary depending on the specific type of beverage (Table 7-1). Although most alcohol is consumed orally, there are isolated cases of individuals injecting ethanol intravenously (1). In addition, ethanol vapor can be inhaled, and machines called AWOL (alcohol without liquid; www.awolusa.com) have been introduced into the United States as a novel means of self-administering alcohol. A number of US states have since banned the sale or use of these devices.
TABLE 7-1 AMOUNT OF ALCOHOL IN DIFFERENT BEVERAGES AND RELATIONSHIP TO A STANDARD DRINKa
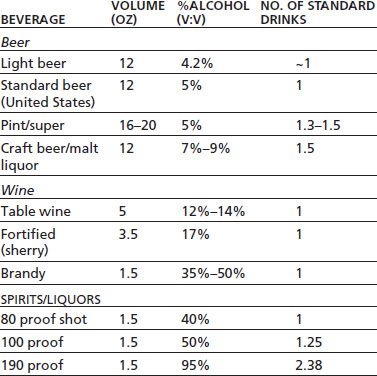
a In the United States, a “standard drink” is one that contains ~0.6 fluid ounces (14 g) of pure alcohol.
In addition to its use as a topical antiseptic, alcohol has several clinical indications including treatment of accidental or voluntary ingestion of methanol or ethylene glycol (2). Ethanol has a higher affinity for alcohol dehydrogenase (ADH) than does methanol and thus reduces the formation of methanol metabolites formaldehyde and formic acid. For both indications, hemodialysis is the recommended first line of treatment. Alcohol has also been used for treatment of various types of cysts (sclerotherapy) and was used historically for treatment of premature labor (2). Safer and more effective medications have largely replaced these uses. Alcohol combined with dextrose is used to increase caloric intake and for replenishing fluids. Alcohol, either given orally or intravenously, is sometimes used by physicians to treat withdrawal, particularly in a hospital setting. For example, a survey of in-patient hospital pharmacies reported that approximately one-half had alcohol available for treatment of withdrawal with surgical services requesting the majority of these uses (3).
Alcohol is one of the oldest used substances. Consumption of alcohol-containing beverages predate recorded human history, while written records of its use are found in Chinese and Middle Eastern texts as far back as 9,000 years ago. Alcoholic beverages have long been consumed as part of the daily diet and are also used for medicinal or symbolic effects. In modern times, alcohol is second only to caffeine in incidence of use, and its manufacture, distribution, and sale are of major economic importance across the world. Alcohol use worldwide is regulated by both societal and religious beliefs. In the United States, consumption of alcohol was legally restricted on the national level with passage of the Volstead Act and the Eighteenth Amendment to the US Constitution in 1919 (4). Amid growing public outcry, prohibition was repealed in 1933 with the ratification of the Twenty-first Amendment that gave states the right to regulate the purchase and sale of alcohol. Today, sales and consumption of alcohol are under control of a wide variety of local and state laws.
The lifetime exposure to alcohol is high with nearly 88% of the US population reporting using alcohol at least once in their lifetime (5). In 2011, current alcohol use (defined as use in the past 30 days) ranged from 3.9% among 12- to 13-year-olds to nearly 70% of 21- to 25-year-olds. Prevalence decreased among older groups although it was nearly 50% among 60- to 64-year-olds (5). Annual alcohol-related costs in terms of lost productivity and health care are estimated at $223 billion (6).
Clinical studies of alcohol abuse and alcoholism have led to the idea that there may be several types of alcohol use disorders, based on the appearance and severity of certain alcohol-related problems (7). Two particularly well-known examples of these classifications are the Type I and II forms proposed by Cloninger et al. and the Type A and B forms proposed by Babor and Caetano (8). Cloninger’s Type I and Babor’s Type A share several similarities including (a) later onset of alcohol-related problems (>25 years old), (b) fewer childhood behavior problems, (c) relatively mild alcohol-related issues with fewer hospitalizations, and (d) lower degree of novelty seeking coupled with a preference toward harm avoidance. Type II and B forms show essentially the opposite characteristics as Type I/A and include (a) familial alcoholism, (b) earlier onset of alcohol-related problems, (c) more incidents of alcohol-related problems or violence, and (d) higher preference for risk taking/novelty seeking.
Absorption and Metabolism
Alcohol is a small, water-soluble molecule that is rapidly and efficiently absorbed into the bloodstream from the stomach, small intestine, and colon. The rate of absorption depends on the gastric emptying time and can be delayed by the presence of food in the small intestine. Once in the bloodstream, alcohol is rapidly distributed throughout the body and gains access to all tissues, including the fetus in pregnant women. Although the relationship between alcohol intake and blood levels is body weight dependent, gender is also important. When body weights are equivalent, women show a 20% to 25% higher blood alcohol level than do men following ingestion of the same amount of alcohol (Table 7-2). This appears to be due primarily to less gastric metabolism of alcohol in women as blood levels are not significantly different when alcohol is administered intravenously (9).
TABLE 7-2 ESTIMATED BLOOD ALCOHOL LEVELS FOLLOWING DRINKING
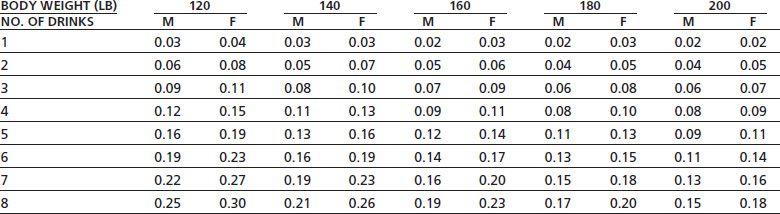
Data represent blood alcohol concentration (gram per 100 mL or gram %) for men (M) and women (F) following consumption of different number of drinks. Subtract 0.01 from each value for each hour of drinking to account for effects of metabolism. 1 drink represents 12 oz beer, 5 oz wine, or 1.5 oz liquor.
Alcohol is metabolized primarily by enzymatic pathways, and only small amounts of alcohol are excreted through the lungs as vapor. The odor of the breath is not a reliable indicator of alcohol consumption, because it is influenced by impurities in the alcoholic beverage. In the liver, alcohol is broken down by ADH and mixed-function oxidases such as P450IIE1 (CYP2E1). Levels of CYP2E1 may be increased in chronic drinkers. ADH converts alcohol to acetaldehyde, which subsequently can be converted to acetate by the actions of acetaldehyde dehydrogenase.
The rate of alcohol metabolism by ADH is relatively constant, as the enzyme is saturated at relatively low blood alcohol levels and thus exhibits zero-order kinetics (constant amount oxidized per unit of time). Alcohol metabolism is proportional to body weight (and probably liver weight) and averages approximately 1 oz of pure alcohol per 3 hours in adults. Thus, the time for an individual to become sober after even moderate intake of alcohol can be substantial. Although stimulants are often used to mask the depressant effects of alcohol, there do not appear to be any truly effective “alcohol antagonists” (amethystic agents) that can quickly reverse the intoxicating effects of alcohol. Naloxone (Narcan), the opiate antagonist, has been tested for its ability to reverse alcohol-induced coma but appears ineffective (10). Several γ-aminobutyric acidA (GABAA) receptor antagonists have also been examined including flumazenil (Anexate) that is used to treat benzodiazepine overdose and the experimental compound RO 15-4513. This compound, which is actually an inverse agonist, has been reported to reverse some of the effects of low to moderate doses of alcohol in animals and may target a very specific subtype of GABAA receptor that shows sensitivity to very low concentration of ethanol (11). In addition to these agents, clinical trials have shown that metadoxine (pyridoxal L-2-pyrrolidone-5-carboxylate) appears to enhance the metabolic clearance of alcohol and speed recovery of intoxicated individuals (12). Although the clinical utility of an antialcohol medication for emergency medicine is undoubtedly high, moral, ethical, and medical concerns must be considered with respect to the development and general use of such a drug since it would not be expected to alter the underlying deleterious effects of alcohol on organ systems.
Central Nervous System
Acutely, alcohol acts as a central nervous system (CNS) depressant. During the initial phase when blood alcohol levels are rising, a period of disinhibition often occurs, and signs of behavioral arousal are common. These include relief of anxiety, increased talkativeness, feelings of confidence and euphoria, and enhanced assertiveness. As drinking continues, there are impairments in judgment and reaction time, increased emotional outbursts, and ataxia. At higher blood levels, alcohol acts as a sedative and hypnotic, although the quality of sleep often is reduced after alcohol intake. In patients with sleep apnea, alcohol increases the frequency and severity of apneic episodes and the resulting hypoxia. Alcohol potentiates the sedative–hypnotic properties of both benzodiazepines and barbiturates, perhaps reflecting common mechanisms of action for these substances. Acute alcohol intoxication is not always associated with sedation or coma; indeed, some intoxicated individuals display violent behavior that requires administration of other sedative or antipsychotic agents. The use of these agents with a severely intoxicated individual must be approached cautiously to prevent respiratory failure. Chronic abuse of alcohol is associated with several psychoses such as Wernicke’s and Korsakoff’s that are associated with deficiencies in nutrition and vitamin intake (particularly thiamine).
Other Organ Systems
An in-depth discussion of the actions of alcohol on various organ systems can be found in other chapters of this book. A brief review of the most common effects is given here. Acute alcohol ingestion usually produces a feeling of warmth as cutaneous blood flow is increased, and this is accompanied by a reduction in core body temperature. Gastric secretions are usually increased, although the concentration of alcohol ingested affects this response, with high concentrations (>20%) inhibiting secretions (13). Long-term ingestion of high concentrations of alcohol can lead to a variety of pathologies associated with the gastrointestinal tract including esophageal varices and bleeding, erosive gastritis, and diarrhea and malabsorption of nutrients and vitamins (13). Alcohol consumption is also associated with an increased risk of tumors in the GI system as well as in other tissues including lung and breast (14). Acute and chronic ingestion of alcohol generally decreases sexual performance in both men and women although sexual behavior may be enhanced due to loss of inhibitory control and judgment. Alcohol has manifold effects on the cardiovascular system, and changes in contractility and function can follow both acute and chronic ingestion of alcohol (15). The deleterious effect of alcohol to the heart and other organ systems is countered by protective effects of small amounts of alcohol on cardiovascular tissue (16). Thus, low to moderate alcohol use is associated with a reduced risk of coronary disease, and this may arise from alcohol-induced changes in plasma lipoproteins and alterations in cell protection pathways. Chronic alcohol ingestion is well known to increase fat accumulation in the liver that can progress to severe liver damage and cirrhosis (17). Alcohol-induced liver damage is due in part to the production of acetaldehyde that readily reacts with proteins, lipids, and other compounds leading to impaired mitochondrial function.
Alcohol has depressant actions on the CNS that are similar to other centrally acting drugs such as barbiturates, benzodiazepines, general anesthetics and solvents, and anti-convulsants. Alcohol also enhances the sedative effects of antihistamines that are commonly used in the treatment of nasal congestion. Combining these medications with alcohol can result in significant CNS depression and reduced ability to safely carry out normal functions such as automobile driving. Alcohol can also enhance the hepatotoxic effects of acetaminophen (Tylenol) and the gastric irritating effects of NSAIDs, thus increasing the risk for development of gastritis and upper GI bleeding. Chronic alcohol use can interfere with the metabolism of certain drugs due to enhanced levels of liver enzymes.
NEUROBIOLOGY (MECHANISMS OF ADDICTION)
A widely accepted tenet of addiction research is that addictive substances, by definition, produce pleasurable effects that engender actions promoting further drug seeking and taking. This concept suggests that drugs of abuse, like alcohol, must produce, at least initially, some form of positive reinforcement that provides a strong incentive to reexperience the drug. As drug use proceeds, the degree of acute reinforcement or reward may be attenuated and drinking escalates as negative aspects associated with withdrawal emerge (18). The repetitive nature of drug use also engages mechanisms of learning leading to entrained behaviors that may become highly ritualistic and habit-like. These findings have led to the idea that drug and alcohol addiction is a form of dysfunctional, maladaptive learning that, once established, is difficult to reverse (19). In terms of brain systems that underlie the development of addiction, recent studies suggest an important interaction between midbrain dopamine-based reward systems and cortical mechanisms of brain plasticity and learning that are mediated by the neurotransmitter glutamate (20).
As reviewed in various chapters in Section 1, all drugs of abuse including alcohol affect reward pathways by enhancing the release of dopamine from midbrain dopaminergic projections that regulate neurotransmission within limbic and cortical circuits that regulate motivated behavior (21). The dopamine (DA) neurons involved in this action reside in the midbrain ventral tegmental area (VTA) and project to discrete areas of the brain, including the nucleus accumbens, olfactory tubercle, frontal cortex, amygdala, and septal/hippocampal areas. These regions are thought to be involved in translating emotion and perception into action through the activation of motor pathways; thus, they may be important in initiating and sustaining drug-seeking behavior. Lesions or inactivation of these discrete brain areas in experimental animals can reduce both the acquisition of drug seeking and its reinstatement following long periods of abstinence (22,23).
The initial reinforcing actions of alcohol appear to involve excitation of VTA dopamine neurons. Acutely, alcohol enhances the firing rate of midbrain DA neurons, and animals will self-administer alcohol directly into the posterior but not anterior VTA (24,25). Chronic exposure to alcohol leads to alterations in the excitability of these neurons in the absence of alcohol that may persist for significant periods of time (26,27). Electrophysiologic studies have demonstrated enhanced efficiency of glutamatergic signaling in neurons following exposure to alcohol and other drugs of abuse (28–30). With respect to reward circuits, these findings suggest that enhanced firing of VTA DA neurons during exposure to alcohol facilitates glutamater-gic transmission in limbic, cortical, and striatal areas, thus strengthening the association between behavioral action and outcome. Studies using in vivo microdialysis to monitor levels of dopamine in freely behaving animals report significant dose-dependent increases in extracellular dopamine levels in the nucleus accumbens in rats self-administrating alcohol (31,32). Importantly, in rats genetically selected to consume large amounts of alcohol, significant increases in dopamine were also observed during the 15-minute waiting period that preceded alcohol self-administration (33). This finding suggests that, in animals with drinking experience, the expected reward that ingestion of alcohol provides is itself sufficient to enhance activity in this pathway. The subsequent pharmacologically induced elevation of dopamine that occurs during drinking may further strengthen the motivation to consume alcohol in future sessions. Genetic differences in the responsiveness of this pathway may contribute to the motivational factors that drive greater alcohol-seeking behavior in certain individuals.
Studies with human alcoholics have examined the neurobehavioral aspects of alcohol abuse, using drug discrimination procedures similar to those used in animal studies. In these studies, human alcoholics are asked to rate the effects produced by a variety of drugs in terms of their similarity to those produced by alcohol. For example, ketamine, a dissociative anesthetic that blocks the ethanol-sensitive N-methyl-D-aspartate (NMDA) subtype of glutamate receptors, induces ethanol-like subjective effects in recently detoxified alcoholics (34). These effects were dose dependent: At low doses, they mimicked the effects of one to two standard drinks of alcohol, whereas higher doses produced effects similar to those of eight to nine drinks. Interestingly, the effects of ketamine that were ethanol-like were associated with the descending phase of blood alcohol concentration that is associated with ethanol-induced sedation.
Other human clinical studies using selective pharma-cologic agents have implicated neurotransmitters such as GABA, serotonin, and the opiates in mediating the rewarding and craving aspects of alcohol action. Results from human imaging studies have begun to identify changes in brain activation during exposure to alcohol or alcohol-related cues between control and alcohol-dependent subjects (35,36). Such human studies are important in the context of understanding the underlying causes of alcohol abuse because alcohol, unlike most drugs of abuse, interacts with a wide variety of molecular and cellular processes to produce its pharmacologic, physiologic, and psychological effects.
Molecular Sites of Alcohol Action
Psychostimulants such as cocaine and amphetamine or opiates like heroin and morphine all produce their primary effect by binding to specific protein receptors expressed on brain neurons. In contrast, alcohol is rather promiscuous and interacts with a wide variety of targets including both lipids and proteins. Initial observations made by the German scientists Meyer and Overton over 100 years ago led to a lipid-disordering hypothesis of alcohol and anesthetic action due to the high correlation between lipid solubility of a compound and its potency as an anesthetic. Support for this theory has waned in recent years due to several factors. For example, measurable changes in membrane fluidity require very high concentrations of alcohol, are relatively modest, and are less marked than the effects produced by small changes in temperature that are not associated with behavioral signs of intoxication (37). These findings and the demonstration of effects in lipid-free systems have led to the idea that alcohol’s acute actions are likely due to effects on ion channel proteins that regulate the excitability of neurons (38). An important point to consider in these in vitro studies of alcohol action is whether the range of concentrations needed to cause significant effects are similar to those associated with the behavioral actions of alcohol. Blood alcohol concentrations in the range of 40 to 400 mg% are equivalent to 8 to 88 mM and are associated with the full spectrum of alcohol intoxication (described in more detail below). Thus, in vitro studies of alcohol’s effect on ion channels are usually conducted at concentrations below 100 mM in order to better approximate levels found in the brain during drinking.
Table 7-3 shows that a variety of ligand-gated ion and voltage-activated ion channels that are expressed by neurons are sensitive to behaviorally relevant concentrations of alcohol.
Alcohol’s acute depressant action on neuronal excitability likely results from its ability to enhance the function of inhibitory ion channels while blocking the activity of excit-atory receptors (Fig. 7-1).
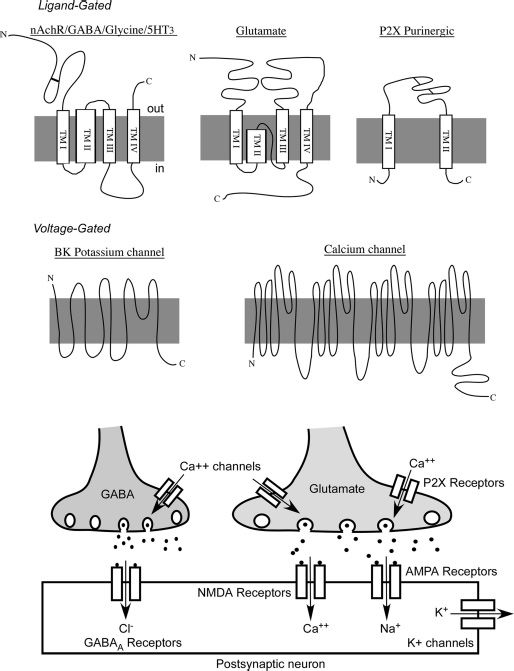
FIGURE 7-1 Structure and location of alcohol-sensitive ion channels in the neuronal synapse.
GABAA and Glycine Receptors
As shown in Table 7-3, there are distinct families of subunits that make up GABAA and glycine receptors. Each class of subunits may have multiple members that differ slightly in their sequence and function. Different subunit combinations give rise to GABAA and glycine receptors that can show variable sensitivity to pharmacologic agents, including alcohol. Alcohol generally enhances GABAA and glycine receptor function, although GABAA ρ receptors are inhibited by alcohol. Initial studies with recombinant GABAA receptors suggested that the γ2L subunit that contains an additional eight amino acids including a site for phosphorylation of the receptor by protein kinase C was important for alcohol potentiation of channel function (39,40). Despite these intriguing results, subsequent studies have shown that γ2L expression is not sufficient in itself to confer sensitivity to alcohol (41). In addition, knockout mice lacking this subunit show normal sensitivity to alcohol, although their responsiveness to benzodiazepines is enhanced (42,43). While these results appear to rule out a major role for the γ2 subunit in mediating alcohol action on GABAA receptors, this may be phosphorylation dependent as the alcohol sensitivity of GABAA α1β2γ2 receptors was reported to be markedly enhanced when the kinase PKCε was inhibited (44). Mice lacking PKCε also show an enhanced response to ethanol suggesting that specific subtypes of GABAA receptors may show appreciable alcohol sensitivity under certain conditions (45).
TABLE 7-3 PROPERTIES OF ALCOHOL-SENSITIVE ION CHANNELS
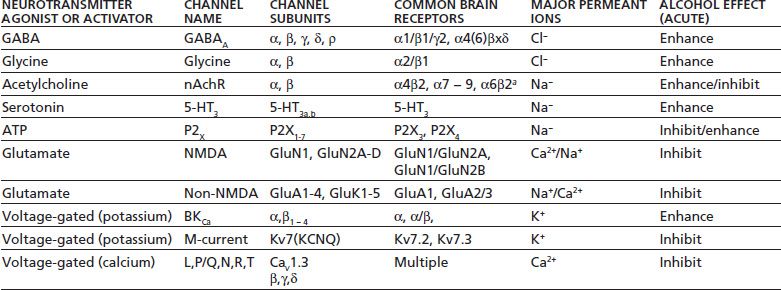
Ion channels are listed according to their natural agonist or mode of activation. Subunit families represent those found in the brain or spinal cord. Brain subtypes are examples of subunit combinations commonly expressed by neurons (aindicates other subunits likely required). Alcohol effect indicates change in ion channel function by alcohol acutely administered to recombinant or native receptor preparations.
In addition to γ-containing GABAA receptors, recent studies suggest that δ-containing GABA receptors possess high sensitivity to relatively low levels of alcohol. These receptors contain the α4(6), β, and δ subunit instead of the γ subunit and are affected by concentrations of ethanol (5 to 10 mM) achieved after only one drink (46). Despite these intriguing findings, not all studies have reported that γ-containing GABAA receptors are sensitive to low concentrations of alcohol (47). While the reasons underlying these conflicting findings are not yet clear, there is substantial evidence to suggest that certain GABAA receptors may be especially important in mediating low-dose effects of alcohol.
A major approach that has revolutionized the study of alcohol action on ion channels is the use of site-directed mutagenesis to alter putative alcohol-sensitive amino acids within a given receptor. These sites are often located within transmembrane domains of individual receptor subunits, and in GABAA or glycine receptors, replacement of a conserved serine by an isoleucine abolished the potentiating effects of alcohol on receptor currents (48). Subsequent studies have shown that this site may also be involved in mediating the effects of some volatile anesthetics on GABAA and glycine receptor function (49). The identification of these alcohol-sensitive residues combined with the development of ion channel structural models has allowed for detailed molecular modeling of these sites and the development of genetically modified animals that express alcohol-insensitive subunits (50).
Interestingly, GABAA receptor knockout and knock-in mice appear to show relatively subtle changes that are restricted to specific alcohol-induced behaviors with little effect on other alcohol actions (51–53). This may reflect the fact that there are multiple targets for alcohol in the brain and that in the case of knockout animals, changes in expression of related GABA subunits may compensate for the loss of those that are alcohol sensitive (54). In addition, there is now good evidence that some of the effects of alcohol on GABA receptor function arise from changes in the presynaptic release of GABA rather than a direct effect on the GABA receptor itself (55). For example, in certain neurons of the amygdala, a brain structure highly involved in processing emotionally relevant stimuli, alcohol’s potentiation of postsynaptic GABAA responses appears to be due to an increase in the release of GABA rather than a direct effect on the channel (56). The mechanism underlying the sensitivity of GABA release to alcohol is not currently known but could involve specific signaling pathways and proteins that normally regulate vesicle movement and fusion (57).
Glutamate-Activated Ion Channels
Glutamate is the major excitatory neurotransmitter in the brain and activates three major subtypes of ion channels called AMPA, kainate, and NMDA receptors, where AMPA is α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid. These channels mediate most of the fast excitatory synaptic transmission in the brain and are critical mediators of most forms of synaptic plasticity thought to underlie learning and memory. NMDA receptors are highly calcium permeable and require both glutamate and glycine for activation, while AMPA and kainate receptors only require glutamate for activation. As reviewed below, NMDA receptors are inhibited by ethanol as well as other abused substances including nitrous oxide, anesthetics, and volatile solvents such as toluene (58–60).
In general, NMDA receptors show appreciable sensitivity to relevant concentrations of alcohol (61), while AMPA and kainate receptors show a more restricted pattern of ethanol sensitivity (62–64). NMDA receptors expressed in most neurons are readily antagonized by alcohol at concentrations (10 to 100 mM) associated with intoxication and sedation although this varies across different brain regions (63–68). Like GABAA and glycine receptors, alcohol inhibition of NMDA receptors may involve an interaction with specific residues within defined transmembrane domains of the receptor that control channel gating (69–71). NMDA responses show a regional and developmental variation in their sensitivity to alcohol, and this may result from differential expression of GluN1 and GluN2 NMDA subunits that regulate some of the alcohol sensitivity of these receptors (72,73). Alcohol’s blockade of excitatory NMDA signaling is likely to be important in mediating some of the acute intoxicating and sedative effects of alcohol (74). Inhibition of NMDA receptors in the prefrontal cortex may underlie some of the cognitive deficits and errors in judgment observed during alcohol intoxication (63,75,76). Long-term alcohol consumption may also affect NMDA receptors in medial and orbital cortical areas that are important for adapting behavior to rapidly changing contingencies (77,78).
The antagonism of NMDA receptors by alcohol may also be involved in its rewarding properties because NMDA receptors are thought to be important in regulating the release of dopamine in mesolimbic areas such as the nucleus accumbens. For example, using microdialysis to monitor changes in dopamine in awake animals, NMDA antagonists were shown to increase levels of dopamine in the nucleus accumbens (79). These results suggest that glutamate, by acting on NMDA receptors on interneurons, may exert an inhibitory control over dopamine release by NMDA receptors. Relief of this inhibition during exposure to alcohol may lead to increases in accumbens dopamine. In animal studies designed to evaluate the subjective effects of ethanol on behavior, NMDA antagonists can produce ethanol-appropriate lever response in rats trained to discriminate ethanol from saline (80).
Since NMDA receptor activity is a critical determinant of normal neuronal activity, it is perhaps not surprising that receptor function is also altered following chronic exposure to alcohol. In primary cultures of neurons, chronic exposure to alcohol increases the density and clustering of NMDA but not non-NMDA receptors (81–83). These changes appear to take place specifically within the gluta-matergic synapse as there is no change in NMDA receptors in nonsynaptic locations. These results suggest that NMDA receptors may serve as alcohol sensors and that neurons compensate for inhibition of these receptors by moving more of them to the synapse where they can be activated by glutamate. One important outcome of this adaptive response is an increased susceptibility of animals and humans to seizures that develop during withdrawal from alcohol. Experiments with mice show that NMDA-induced seizure activity is elevated in mice made dependent on alcohol and that NMDA antagonists reduce or prevent seizures during withdrawal (84). Results from studies using isolated cells or intact animals also suggest that the enhancement in NMDA receptor function after chronic alcohol may involve changes in the expression pattern of specific NMDA receptor subunits (77,85–88).
Other Ion Channel Subtypes
5-HT3 Receptors
5-HT3 receptors are ligand-gated ion channels activated by serotonin. They are permeable primarily to monova-lent cations, such as sodium and potassium. In cultured neurons and recombinant expression systems, low doses of alcohol appear to potentiate currents carried by 5-HT3 receptor (89,90). In behavioral studies involving both rats and pigeons, 5-HT3 receptor antagonists blocked the animal’s ability to discriminate ethanol from saline, suggesting that alcohol’s acute actions on 5-HT3 receptors may underlie some of the subjective effects of alcohol (91).Human studies using the 5-HT3 antagonist ondansetron (Zofran) have generally found that the drug reduces drinking although this effect appears more efficacious in early-onset alcoholics (92).
Acetylcholine Nicotinic Receptors
Acetylcholine activates a family of ligand-gated ion channels expressed in brain neurons that are related to the nicotinic receptor expressed at the neuromuscular junction (93). Alcohol has been shown to either potentiate or inhibit acetylcholine receptors expressed in cultured neurons or oocytes (94,95). In neurons, this biphasic effect appears to result from expression of different subtypes of nicotinic receptors that show a differential response to ethanol. Thus, heteromeric nicotinic receptors composed of just αβ subunits appear to be potentiated by ethanol, whereas homomeric receptors composed of just α subunits (e.g., α7) are inhibited by ethanol (95). It is not yet clear how these different effects of ethanol on neuronal nicotinic receptors are manifested at the behavioral level. However, nicotinic receptors are expressed by neurons in the VTA, nucleus accumbens, and prefrontal cortex where they help shape the excitability of these neurons. Varenicline (Chantix), a medication approved for smoking cessation, is a partial agonist at the α4β2 nicotinic receptor. Results from preclinical studies have reported that varenicline also reduces alcohol seeking in rodents without altering responding for sucrose (96).
ATP-Gated Ion Channels
Adenosine triphosphate (ATP) is released into synapses where it acts on ATP-gated ion channels of the P2X family (97). Alcohol inhibits ATP-gated channels in some neurons (98), while the effect of alcohol on recombinant P2X receptors is subtype specific. P2X2 and P2X4 channels are inhibited by alcohol (98–100), but currents from P2X3 receptors are enhanced (101). This appears to involve differences in a small number of amino acids between subunits at sites near the transmembrane domains that control channel gating (102). The inhibitory action of ethanol on the P2X4 receptor is blocked by the anthelmintic drug ivermectin that is a positive allosteric modulator of P2X4 channel function (103), and in mice, ivermectin reduces ethanol consumption in a variety of drinking models (104).
Potassium- and Calcium-Selective Ion Channels
Potassium channels that are regulated by calcium (SK and BK channels) and those gated by G proteins (GirK) channels are also affected either directly or indirectly by alcohol (105). Potassium channels serve as a brake on excitatory glutamatergic transmission by hyperpolarizing the membrane and thus are critical regulators of neuronal activity. BK channel activity is acutely enhanced by alcohol, and this enhancement may contribute to the inhibition of vasopressin release from neurohypophysial terminals and the resulting diuresis that accompanies alcohol ingestion (105). SK channels do not appear to be inhibited directly by ethanol, but their expression and localization in hippocampal neurons are altered following chronic exposure (106), and these changes may contribute to ethanol withdrawal hyperexcitability. In addition, down-regulation of SK channels in nucleus accumbens following chronic alcohol exposure may contribute to elevated drinking as enhancing SK activity with an allosteric modulator (chlorzoxazone; Parafon Forte) reduced alcohol consumption in rats (107). Most voltage-gated potassium and sodium channels are relatively insensitive to relevant concentrations of alcohol although there are exceptions. For example, recombinant Kv7 (previously termed KCNQ) channels are inhibited by moderate concentrations of ethanol (108), and Kv7.2/Kv7.3 heteromers are thought to underlie the slow, noninactivating M-current that reduces neuronal excitability. In VTA DA neurons, M-current is inhibited by acute ethanol at concentrations that enhance DA neuron firing suggesting another possible mechanism for ethanol-induced increases in DA release (109). Alcohol also has inhibitory actions on certain subtypes of voltage-gated calcium channels (110), and genetically modified mice lacking the N-type calcium channel show reduced ethanol consumption as compared to wild-type animals (111). Alterations in T-type calcium channels in thalamic neurons by alcohol may contribute to disruptions in sleep that are commonly observed in alcohol-dependent individuals (112).
Pharmacologic Studies Implicating Other Neurotransmitter Systems
Much of the working knowledge of alcohol’s effects on neuronal function comes from animal studies examining the effects of neurotransmitter-specific agents on alcohol drinking. A brief review of this literature is presented here and is summarized in Table 7-4.
TABLE 7-4 EFFECTS OF ACUTE AND CHRONIC ALCOHOL ON VARIOUS BRAIN ION CHANNELS AND NEUROTRANSMITTER SIGNALING SYSTEMS
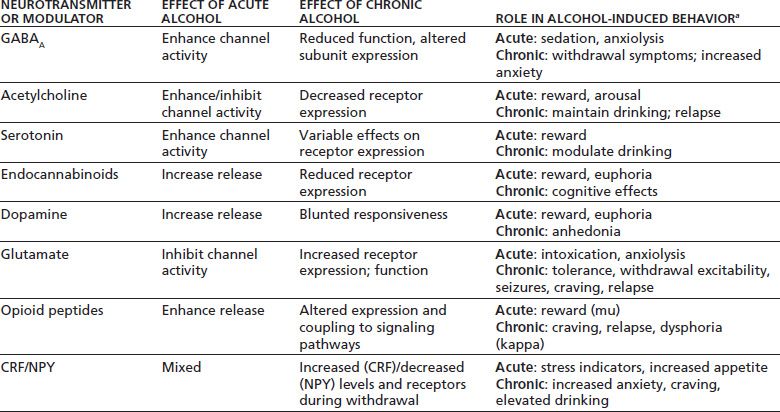
aBehaviors that each system/neurotransmitter may contribute to.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


