93
CHAPTER OUTLINE
Chronic pain is our society’s most common and most costly chronic illness (1,2). It has the potential to change a person’s life trajectory, sometimes catastrophically, when treated inadequately or unwisely. Too often, the desperation of patients with pain leads them to be particularly vulnerable to the promise of cure or relief from the marketing of new products and procedures that lack an evidence basis for effectiveness. Opioid analgesics pose a particular dilemma. With their incredible capacity to ease the suffering of pain, they strongly impact the reward systems in the brain that play a key role in the activation and perpetuation of addiction. Thus, the two together—chronic pain and addiction—present a “perfect storm” of complexity and a challenge to our health care system and to society at large. Patients, their families, their employers, their communities, and their health care providers share the burden of chronic pain, contribute to the perpetuation of unwise and ineffective policies and procedures, and have a role to play in improving the outcomes of chronic pain and the outcomes of addiction, particularly when they complicate each other. For purposes of this chapter, we will not be considering cancer pain caused by malignant cells that damage tissues and the nervous system; rather, we will be referring to the cluster of noncancer conditions and diseases associated with persistent pain (e.g., musculoskeletal conditions, neurologic damage) that share common neuropathophysiologic mechanisms, using the term “maldynia” (denoting “bad pain” or “pain as a disease”) as distinct from eudynia (normal, acute pain in response to threatened or actual tissue damage or injury). Unraveling the biopsychosocial factors that perpetuate maldynia and impact a person’s pain control and addressing these factors in an organized, prioritized, goal-oriented management plan challenge our understanding of behavioral neuroscience, our use of available medical and team-based skills, our capacity for empathy, and our patience and perseverance (3,4). This challenge extends to the optimization of our health care system so that it incentivizes outcomes instead of profit (5–7). The pathophysiology of maldynia does not alone account for its variable symptomatic and behavioral expression and psychosocial complexity. Neural and glial networks regulating pain perception and behavior interact with emotional regulation and reward systems influencing addiction. Physiologic responses in both are worthy of examination with the expectation that the coexistence of maldynia and addiction will lead to complex interactions, that is, addictive responses altered by maldynia and maldynia altered by addiction.
This chapter reviews the pathophysiology of maldynia and the theoretical and clinical evidence for its overlap with addiction. The mechanisms of pain perception and modulation in maldynia are reviewed, with discussion of those aspects likely to be impacted by the presence of addictive disease. In this regard, the role of central sensitization and kindling and affective responses including depression and anxiety in maldynia is discussed, particularly since these processes and comorbidities also play a role in the phenomenology and treatment of addiction. Finally, ideas about potential interactions between maldynia and addiction are suggested, focusing on how the general state of addiction, inherited characteristics of the individual, and neuroadaptations specific to opioid abuse might affect the coexpression of these phenomena.
CHRONIC PAIN
In contrast to our ability to prevent, minimize, and manage acute pain, the management of maldynia often presents a daunting challenge. First, the physiology of pain after initial onset becomes much more complex almost immediately— the longer the pain, the more complex the process. The concept of “chronification” increasingly supplants the older, less useful, dimensional division between acute and chronic pain (8,9). Pain is now more accurately described as a continuum of experience from the moment of its onset until its cessation, and that experience informs the next experience of pain, through the glial cellular and neuronal network memory systems. As Emily Dickenson so prophetically wrote 150 plus years ago,
Pain has an element of blank.
It knows not when it began,
Or if there was a day when it was not.
Its infinite realms contain its past,
Enlightened to perceive new periods of pain.
Chronic nociceptive pain, such as from arthritis, involves processes that include transduction/activation, transmission, modulation/augmentation, and perception, whereas chronic neuropathic pain, caused by damage to neural tissue, is often independent of transduction and involves persistent activation, transmission, modulation/augmentation, and perception. These processes will be discussed below in more detail. In both types of persistent pain, a number of biopsychosocial factors can potentially influence neuroplastic changes in the central nervous system (CNS) that will, in turn, determine the salience of the pain for a person, the impact of pain on their lives, and pain’s resistance to treatments that focus on the nociceptive stimulus (10–12). These factors include, but are not limited to, environmental context, meaning of pain, attention, mood and affect, anxiety, catastrophizing, and social and cognitive attention/reinforcement.
Although experiments using laboratory-induced nociception in animals can control for much genetic and experiential variability, this is not possible in humans suffering chronic or episodic pain. Every new episode or change in pain intensity, character, or localization activates neural and inflammatory mechanisms informed by prior nociceptive experience and cognitive–emotional processes that are influenced by current context and meaning conditioned by past experience. These processes involve inflammatory glial activating systems in the central nervous system interacting with neural networks subserved by a myriad of chemical messengers that communicate among sensory systems and various cognitive–emotional processing and behavioral systems (13,14).
Pain is always an intensely personal and unique experience. However, pain experiences do cluster in specific, predictable patterns that can be diagnostically informative, for example, the pattern of pain can be nearly diagnostic in such conditions as S1 radiculopathy, migraine, fibromyalgia, diabetic neuropathy, and postherpetic neuralgia. Clinicians caring for patients with maldynia face the challenge of formulating for each patient, to the degree possible in a particular clinical setting, the interaction of biopsychosocial factors and neurobehavioral processes that activate and perpetuate chronic pain. Then they must devise a feasible treatment plan that has the best chance of remediating the most salient factors to improve the longitudinal outcome (3,5).
Pain Anatomy and Physiology
Wall and Melzak’s (1965) (15) postulation of the gate theory of pain provided the first pathophysiologic model to coherently explain the phenomenology of pain perception and modulation that was observed clinically. One author’s (RG) first memorable clinical experience illustrating the “gating system” occurred when delivering babies in a rural family practice setting in the early 1970s. During training, he had witnessed childbirths in an urban city hospital, perceived by the mothers as culturally foreign and threatening, since often they had received little prenatal care. These women usually experienced severe pain, high anxiety, and almost always required systemic opioids and neural blockade during labor and delivery. In contrast, his rural patients, when provided prenatal care by their own family doctors in a community setting and supported by training in Lamaze techniques, would usually give birth without systemic analgesia, despite the same tissue injury and volleys of nociception inherent to the process. In many cases, at their preference, they delivered without regional blocks. This suggested that other factors such as psychological state and its physiologic effects somehow affected the transmission of pain sensation and its experiential severity. Other examples abound of the phenomenon of environmental conditions and psychological state altering pain experience following normally very painful nociceptive injury—such as injured athletes not experiencing pain during competition or wounded soldiers not experiencing pain during battle.
Like any theory explaining the phenomena of great salience, the gate theory initiated an explosion of pain science, inspiring a generation of clinical and basic scientists who have dramatically expanded our knowledge about the nature of pain physiology, pathology, and experience. A review of the number of pain-relevant articles retrieved in PubMed using keyword indicators “pain” and “nociception” reveals about a twofold increase in publications containing these terms in the title or abstract, with over 160,000 published in the last decade. This new science has radically changed our understanding of the pathophysiology of chronic pain conditions and has generated a plethora of new treatments targeted at different molecular and neurobehavioral mechanisms affecting pain sensation. Interest in clinical pain has similarly exploded. A similar search reveals dramatically increased penetration of the science of pain into the general medical literature. Clearly, pain science and pain management are no longer the province of just pain specialists and scientists; they have penetrated the collective consciousness of our clinical medical culture.
Naturally, this new knowledge about the complex mechanisms involved in the experience of pain has revealed multiple targets for treatments. Some aim to reduce the stimulus itself—for example, weight loss to reduce weight-bearing stress on injured or diseased joints in the lower limbs or lower spine or postural and ergonomics training in musculoskeletal diseases leading to less “transduction” (activation of nociceptors). Others aim to block the stimulus from reaching the spinal cord (transmission), for example, peripheral nerve blocks, or aim to deactivate the transmission of the pain signal in the spinal cord rostrally to the brain (transmission), for example, neuromodulation. And still others target central nervous system responses and pathologic neuroplasticity—for example, neural reprogramming using cognitive–behavioral and psychomotor trainings to improve coping and reverse pathologic neuroplastic changes (16–19). In the hopes of positively affecting all of these processes, clinicians commonly combine treatments, each targeting different mechanisms in the pathophysiologic matrix of maldynia (3,4,10,12,20–22).
This additive, integrated approach improves pain control and outcomes as demonstrated by meta-analytic studies of comprehensive rehabilitation center populations (23–26). Although scientific comparisons of a single proven treatment with combinations of two or more proven treatments are prohibitively expensive, multimodality treatment to improve functional outcomes, not just pain control, is relatively well established as the most effective and longest-lasting treatment for chronic non-cancer pain (CNCP).
The neurobiologic basis for Wall and Melzack’s functional gating system, now better understood, provides a conceptual framework for understanding the clinical presentation of pain and helps explain the often complementary effects of different treatments for pain. For convenience, the very complex phenomenology of pain can be characterized as three major “stages” involving different mechanisms depending on the nature and time course of the originating stimulus (27,28). The overall anatomy of the systems for pain perception and transmission is shown in Figure 93-1. The stages are (a) nociception, the consequences of a brief noxious stimulus; (b) persistent and/or repeated nociception, the consequences of a prolonged or repetitive noxious stimulus leading to tissue damage and peripheral inflammation; and (c) the consequences of neurologic damage, including peripheral neuropathies and peripheral and central sensitization. These stages are not mutually exclusive and overlap temporally and physiologically through the complex interaction among various brain regions and ascending and descending messages between the CNS and the body. Their end result is chronic pain perpetuated by one or more of several mechanisms: chronic inflammation or recurrent injury peripherally; ectopic excitability of peripheral pain neurons; excitation and priming of spinal microglial cells; structural reorganization (e.g., neuroplasticity) of neural networks in the periphery and central nervous system so that touch, movement, temperature, and emotional changes cause the sensation of pain (discussed later—see Table 93-1); and loss of descending inhibitory controls. At any given time, one or a combination of these pathophysiologic mechanisms may be contributing to the experience of maldynia. Figures 93-2 and 93-3 pictorially represents this cycle and recycle of neurophysiologic events and biopsychosocial phenomenology leading to pain’s sometimes terrible consequences that are captured by the construct of maldynia.
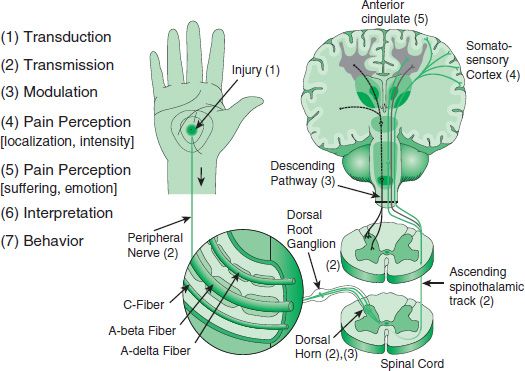
FIGURE 93-1 Anatomy of pain perception.
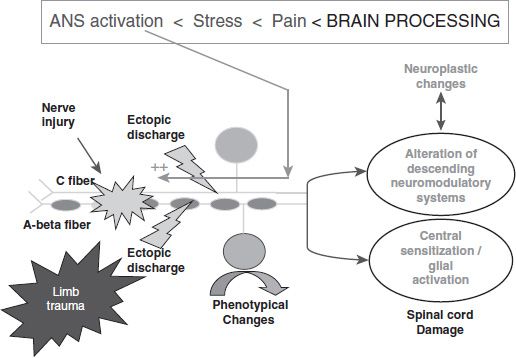
FIGURE 93-2 Key: After injury to first-order nociceptive fibers, they spontaneously and repetitively discharge, activating the biochemical cascade, leading to central sensitization and neuroplasticity. Pain activates the stress response, including the sympathetic system, which can also be activated by emotional states such as anxiety or frustration not directly related to pain. Sympathetic terminals on injured nerves and/or nociceptors release norepinephrine, respectively, increasing ectopic discharge of injured nerves and/or lowering nociceptor firing threshold in injured tissue. This pathophysiologic process increases pain such that commonly patients with maldynia are known to “hole up” socially, avoiding interpersonal contact and other situations that might aggravate them and worsen their pain. (Adapted from Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 1999;353(9168):1959–1964; Attal N, Bouhassira D. Mechanisms of pain in peripheral neuropathy. Acta Neurol Scand Suppl 1999;173:12–24; discussion 48–52.
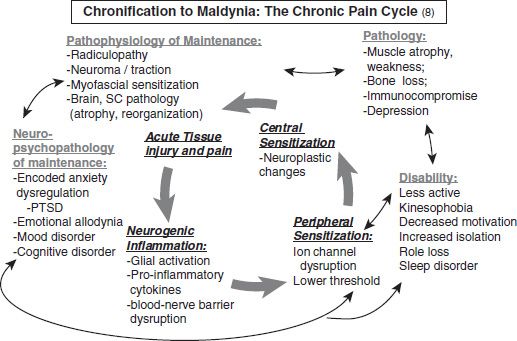
FIGURE 93-3 Acute to chronic pain cycle. (From Gallagher RM. Pharmacological approaches to pain management. In: Ebert M, Kerns R, eds. Behavioral and psychopharmacological pain management. New York, NY: Cambridge University Press, 2010:129–141.)
TABLE 93-1 SIMPLE BEDSIDE EXAMINATION FINDINGS AND THEIR MEANING
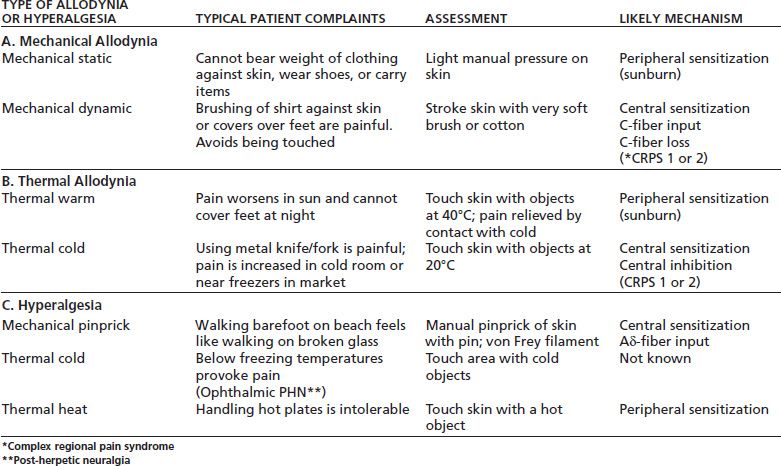
*Complex regional pain syndrome
**Post-herpetic neuralgia
Stage 1: Nociception
A sufficiently strong noxious stimulus (mechanical, thermal, or chemical) activates nociceptors leading to depolarization of pain afferents (A delta and C fibers) that transmit the pain message from the peripheral tissue to the dorsal horn of the spinal cord. If the signal is strong enough (many nociceptive neurons firing) and/or repetitive enough, second-order neurons depolarize, sending the message rostrally to the lateral and medial thalamus. There, projections to the somatosensory cortex convey localization and intensity information resulting in the conscious perception of pain. Projections from the medial thalamus to the limbic system, specifically the anterior cingulate cortex and insular cortex, activate suffering and the emotional aspect of pain, as in Figure 93-1. Once this entire system is activated, repetitive nociceptor stimulation in the periphery can trigger a hyperresponse of the second-order neuron, increasing pain despite no increase in nociceptor input.
Stage 2: Peripheral Sensitization
The situation changes in Stage 2 pain when a noxious stimulus is very intense or prolonged leading to tissue damage and inflammation. Many nociceptors are inactive and unresponsive under normal circumstances; however, cell damage and death from injury or disease and accompanying inflammation can cause an “awakening” of nociceptors, which may spontaneously discharge and become more sensitive to peripheral stimulation (27,29). This is one type of peripheral sensitization. The sensitization of nociceptors occurs when such inflammatory mediators as bradykinin, prostaglandins, serotonin, and histamine (30) are released in damaged tissue. They bind to specific receptors on nociceptive afferents, which elicits the activation of second messenger systems. Ultimately, this leads to phosphorylation of receptors, influx of Ca2+ ions (which further sensitizes the cell), and release of such chemicals as substance P, which promote continued release of inflammatory mediators. Several types of pharmacologic receptors, including opioid, γ-aminobutyric acid (GABA), bradykinin, histamine, serotonin, and, more recently, capsaicin (31), have also been identified on the surface membrane of sensory axons.
The consequence of peripheral sensitization is that depolarization of primary afferent pain fibers now occurs following stimuli that previously would have been subthreshold or even spontaneous, that is, sensitized nociceptors may fire without a noxious stimulus being present. The firing of an increased number of nociceptive afferents codes for an increase in the pain signal to the spinal cord and brain, causing increased pain from a given noxious stimulus—this is termed hyperalgesia.
Also in this circumstance, pain can result from normally innocuous light touch that activates sensitized peripheral neurons (32)—this is termed allodynia. A sunburn provides a good clinical example of a temporary state of peripheral sensitization, whereas arthritis would be a prolonged, chronic state as in Table 93-1.
Along with nociceptor sensitization, changes take place in the central pathways. The central pathways for processing nociceptive information begin at the level of the spinal cord (and medullary) dorsal horn. The interneuronal networks in the dorsal horn transmit nociceptive information not only to neurons that project to the brain but also to spinal motor neurons, which leads to enhanced reflex actions, including muscle spasm, in response to a noxious stimuli. Other inputs result in inhibition of projection neurons. The balance of these excitatory and inhibitory processes provides an explanatory basis for gate theory of pain transmission (33).
Following damage to peripheral nerves, whether by neurologic disease, amputations, surgery, radiation, or chemotherapy, pain fibers can fire spontaneously, can be triggered by trivial stimuli, and can fire in response to adrenergic outflow (such as occurs with psychological stress). Continual firing can lead to changes in the spinal cord including alterations of modulatory systems, central sensitization, and neuroplasticity, whereby dendritic formation in polymodal afferent fibers contributes to pain sensation, causing allodynia. These processes are illustrated by Figure 93-2 (34,35).
State 3: Central Sensitization
In central sensitization, persistent noxious input sets off a process of enhancement of responsiveness in the dorsal horn neurons that continues independent of primary afferent drive. Central sensitization is thought to be responsible for the clinical presentation of secondary hyperalgesia (hyperalgesia in sites away from the site of initial injury) and allodynia. The neurotransmitters contained in the terminals of nociceptive afferents in the dorsal horn include excitatory amino acids, particularly glutamate, as well as neuropeptides such as substance P, calcitonin gene–related peptide, vasoactive intestinal peptide, somatostatin, and others. The rationale for preemptive analgesia, when analgesics and/or neural blocks are given prior to a surgical procedure, is that such interventions are thought to prevent central sensitization (36). Inhibition of nociceptive circuits, mediated by a number of neurotransmitters, including amino acids such as 5-hydroxytryptamine, GABA, and glycine as well as neuropeptides such as enkephalins, forms the basis of many pharmacologic interventions.
Stage 3 pain states are abnormal pain states, chronic pain conditions that are often grouped as maldynia (bad pain). This term was proposed to better encompass not only the neuropathic pain (primary maldynia) initiated or caused by a primary lesion or dysfunction in the nervous systems but also the pain resulting results from inadequately relieved persistent nociceptive stimulation, also defined as secondary maldynia (37).
These chronic pain states are generally the consequence of chronic inflammation secondary to structure injury (e.g., arthritis), damage to peripheral nerves (e.g., neuropathy), damage to the CNS (e.g., thalamic stroke, multiple sclerosis), or changes in the CNS itself, for example, neuroplasticity, central sensitization, and alteration in modulatory systems like descending inhibitory systems (e.g., complex regional pain syndrome, as in Fig. 93-2). Maldynia is characterized by a lack of correlation between the intensity of a peripheral stimulus and the intensity of pain (manifest by exaggerated responses to noxious stimuli) and also by pain that is spontaneous or triggered by innocuous physical or psychological stimuli (38).
Descending control from supraspinal centers inhibits ascending nociceptive transmission, and this control is proportional to the afferent nociceptive input by C fibers (35).
It has been suggested that loss of a proportion of the normal input after peripheral nerve damage produces a lifting of descending inhibition, which could be responsible for enhanced pain perception, particularly for the phenomenon of hyperalgesia, in which there is an exaggerated pain response to a noxious stimulus (27).
However, the development of stage 3 pain may also involve genetic, cognitive, and emotional factors that remain to be clarified (33,39).
Recent brain imaging studies suggest that more rostral changes in the CNS also mediate sensitization. For example, changes in the hippocampus may sustain potentiation of an initial stimulus for up to a year, which can be reversed by placing laboratory mammals in a challenging novel environment (40). This finding supports a widely held clinical belief that stimulating, goal-directed activity and exercise suppress pain, enable improvements in functional ability, and even reduce the manifestations of central sensitization (e.g., allodynia and hyperpathia). This phenomenon is thought to be one reason for the effectiveness of comprehensive rehabilitation programs in treating chronic pain. Regions in the cingulate gyrus have now been identified that are responsible for modulating behavioral responses to pain, attention to pain, pain induced fear, and learning and pain (41–45). Figure 93-3 illustrates the sequence of biopsychosocial processes involved in the transition from acute injury to maldynia and its related comorbidities and disability. Treatment hence involves a comprehensive approach that identifies the patient’s biopsychosocial profile in this phenomenologic pathway and addresses specific pathophysiologic factors that perpetuate pain and pain-related and disease-related impairments and comorbidities (see Fig. 93-3).
Pain and Emotions
Emotional states that activate sympathetic arousal, such as anxiety or anger, can increase acute pain and reactivate or worsen chronic pain. When depression and anxiety disorders, such as posttraumatic stress disorder (PTSD) and maldynias, are comorbid, they complicate each other’s treatment, such that treating one condition without treating the other effectively increases the likelihood of treatment failure. Moreover, certain personal traits, such as external locus of control (46,47) and a tendency to catastrophize (48–50), also predict worse outcomes. Finally, comorbid substance abuse may complicate the use of opioid analgesics for pain control. Substance abuse, by interfering with the sustained, goal-oriented behavior needed for functional restoration, leads to poor outcomes of pain treatment generally. The mechanisms for this effect will be discussed in more detail below. Thus, identifying and managing these comorbidities are critical to effectively treating pain.
Historical Notes on Psychosomatic Concepts
A review of rigorous epidemiologic studies over several decades demonstrates a strong association between depressive and anxiety disorders and chronic pain (50,51). Although chronic pain generally affects psychological health and functional status consistently across cultures (52), the relationship is more complex when considering specific painful diseases. For example, a mechanism for the high association between depression, anxiety disorders, and arthritis is suggested by recent studies linking the presence of depression to an impaired capacity to regulate the inflammatory cascade during stress (53).
The causal direction of the relationships among chronic pain disorders and psychiatric comorbidities and the relative influence of environmental factors and genetic/familial factors, “nature versus nurture,” are also being studied. For example, a series of studies of patients with myofascial facial pain demonstrated that major depression was more common than in community controls (54), that pain and depressed mood seasonally covaried (55), and, using a family study methodology, that the stress of living with chronic pain increased the risk for depression in persons without a personal or family history of depression (56). This latter study suggests that the stress of living with pain, not familial risk, is the dominant cause of comorbid depression in chronic facial pain. However, using a similarly rigorous methodology, a series of family studies of community-dwelling adult females with a different pain disease, fibromyalgia, a disorder marked by generalized muscle pain and tenderness with no peripheral pathology, supports the hypothesis that fibromyalgia is a depressive spectrum disorder (57). This and other studies (58) suggest a shared pathogenesis, such as dysfunction in serotonin and norepinephrine systems, in fibromyalgia (59–62), and in depression (63,64). Shared genetic risk factors for fibromyalgia and major depressive disorder may also be identified, for example, genetic polymorphisms in serotonin-related genes in fibromyalgia (65–68). Thus, the increase in rates of major depression in families of persons with fibromyalgia, with or without depression, may reflect vulnerability in these neurotransmitter systems that influence both disorders.
The effects of environmental factors on the pathogenesis of pain disorders and depressive disorders may be shared. Stress and trauma, implicated in both facial pain (56) and depression (69–72), may alter the course of pain in subjects with existing pain. In a prospective study of a community sample of fibromyalgia and major life trauma (the 9/11 World Trade Center attack), exposure to stress was not associated with an increase in rates of fibromyalgia in controls (community subjects without fibromyalgia) but was associated with increases in pain in community subjects with fibromyalgia already confirmed by research diagnostic criteria (73).
One interpretation of these aforementioned studies is that central states such as fibromyalgia and major depressive disorder (MDD) share a genetic or other biologically mediated vulnerability to respond to stressful or traumatic events with psychological and pain-related symptoms. This interpretation is consistent with two recent studies, one showing that persons with fibromyalgia symptoms are more likely than community controls to develop PTSD in response to a fixed level of exposure to traumatic events
(73) and another demonstrating that during experimental interpersonal stress, women with fibromyalgia who were primed to experience negative mood prior to stress showed greater subsequent pain elevations than similarly primed women with osteoarthritis (74). Genetic studies suggest that a functional polymorphism in the promoter region of the serotonin transporter gene affects the influence of stressful life events on depression (75), a region that has also been implicated in some investigations of fibromyalgia (66,67).
Pain and Anxiety
The experimental and epidemiologic evidence points to a strong association between anxiety and pain and anxiety disorders and pain (50,51) mediated in part by the amygdala, which appears to play a key role in the regulation of the emotional response to pain. The “nociceptive amygdala” can be influenced by a wide range of environmental and internal stimuli to modulate the subjective experience of pain. The hippocampus, a structure with robust connections to the amygdala, is a center for memory formation, storage, and retrieval. It provides information from detailed memories that are processed by the amygdala and given a particular emotional value, which may influence the development of “pain memory” after central sensitization (40). The emotional value the amygdala places on a particular memory is then fed back to the hippocampus, which integrates this information and either strengthens or weakens the memory. This is why it is believed that events associated with high emotional content, and often painful injury, such as a car accident or being wounded in battle, tend to be remembered in greater detail, as observed in PTSD, than those experiences with little emotional significance (back pain on your drive to work every morning or working in the garden on the weekend). Exposure to novel, “interesting” environments can reverse hippocampal sensitization in pain experiments (40). Studies of disorders such as complex regional pain syndrome (formerly called reflex sympathetic dystrophy or RSD) show that specific sequences of mental activities (16,76) and biofeedback using brain imaging (17) reduce pain. These studies support the usefulness of rehabilitative approaches emphasizing patient involvement in goal-oriented, motivating, engrossing activities (18,19).
PTSD, which is often comorbid with chronic pain associated with physical injury, is associated with changes in the amygdala, hippocampus, and other areas of the limbic system (the neural circuits of complex emotional experience). Neuroimaging studies have found consistent reductions in either total hippocampal volume or blood flow in men and women with PTSD (77–80). influence the amygdala. Human imaging studies have found that the fusiform gyrus, prefrontal gyrus, and anterior cingulate gyrus are preferentially activated in response to fearful stimuli (81,82). The orbitofrontal cortex (OFC), which is involved in the evaluation of risk and reward and social norms, may also have a direct role in regulation of anxiety via its connection to the amygdala (83). The cortex, then, plays an essential role in the categorization, appraisal, and attenuation of our reactions to fearful stimuli, such as pain. These connections are outlined in Figure 93-4. The higher cortical connections to the more primitive fight, flight, and reward circuitry provide a degree of conscious recognition and control over these processes. These connections and their conditioning suggest the biologic basis for the effects of behavioral treatments used widely in chronic pain, such as relaxation and biofeedback. They also suggest specific targets for neuromodulation.
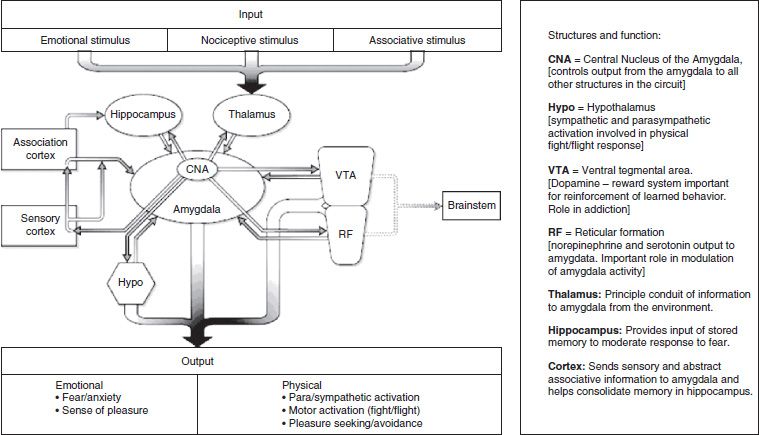
FIGURE 93-4 The circuitry of fear. (From Tirado CF, Gallagher RM. The diagnosis of anxiety and anxiety disorders in chronic spinal pain. In: Slipman C, Wetzel T eds. Interventional spine: an algorithmic approach. New York: Elsevier Health Sciences, 2007.)
More recent functional brain chemistry research has provided neuroanatomical evidence for the overlap between the processing and perception of pain and anxiety. In an experiment comparing patients with chronic low back pain (CLBP) to normal controls, significant differences were found in two regions of the association cortex (orbitofrontal [OFC] and dorsolateral prefrontal cortex [DLPFC]), the cingulate gyrus (part of the limbic system), and the thalamus (84). The study showed that persons with CLBP have differences in regional brain chemistry in the OFC and DLPFC when compared with those without pain. Additionally, persons with CLBP and anxiety had changes in chemistry suggesting increased interaction among all four brain regions, whereas anxious controls only had changes observed in the OFC. Anxiety and pain, therefore, share common neurochemical pathways and can interact in a way that leads to the reorganization of perceptual pathways in the brain. Living with chronic pain is stressful and is thought to be associated with physiologic and psychological changes, yet there is a knowledge gap regarding brain elements involved in such conditions. Baliki and Apkarian’s studies with fMRI suggested that the subjective spontaneous pain of chronic back pain involves specific spatiotemporal neuronal mechanisms, distinct from those observed for acute experimental pain, implicating a salient role for the emotional brain concerning the self (85).
CLINICAL INTERFACE BETWEEN PAIN AND ADDICTION
Multiple points of interface exist between pain and addiction, and these become significant at the clinical level. Pain and drug reward share common neuroanatomical and neurochemical substrates, and the physiologic sequela of addiction (i.e., tolerance, dependence, altered stress response) have clear effects on pain management. Drugs of abuse often have analgesic properties, yet the disease of addiction brings with it malaise, mood states, behaviors, and social losses that serve to worsen the pain experience. The clinical interaction of pain and addiction is particularly complex in the case of addiction to opioids, as these drugs appear to be imbued with both analgesic and hyperalgesic properties. Further, accumulating genetic data suggest that pain, opioid analgesia, and opioid addiction may share similar patterns of gene expression, which become evident in the response of the individual patient.
Neurobiologic Mechanisms of Addiction
Like pain, addiction is an extremely complex human response, with strong behavioral components, that cannot be entirely understood yet by analyzing its physiology. Addiction can be defined as a chronic, relapsing disorder that has been characterized by (a) a compulsion to seek and take drugs, (b) loss of control over drug intake, and (c) emergence of a negative emotional state (e.g., dysphoria, anxiety, and irritability) that defines a motivational withdrawal syndrome when access to the drug is prevented (86). The occasional, limited, recreational use of a drug is clinically distinct from the loss of control over drug intake and the emergence of compulsive drug-seeking behavior that characterize addiction.
Addiction has been conceptualized as a three-stage cycle—binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation—that worsens over time and involves dramatic changes in the brain reward and stress systems (87). Two primary sources of reinforcement, positive and negative reinforcement, have been hypothesized to play a role in the development of addiction. Positive reinforcement is defined as the process by which presentation of a stimulus increases the probability of a response; negative reinforcement is defined as the process by which removal of an aversive stimulus (or aversive state in the case of addiction) increases the probability of a response. Different theoretical perspectives from experimental psychology (positive and negative reinforcement frameworks), social psychology (self-regulation failure framework), and neurobiology (counteradaptation and sensitization frameworks) can be superimposed on the stages of the addiction cycle (86). These stages are thought to feed into each other, become more intense, and ultimately lead to the pathologic state known as addiction. Standard diagnostic criteria for addictive disorders (DSM-IV-TR) (88) not only rely on the more overt physiologic responses to chronic drug use (two of seven criteria such as tolerance and withdrawal) but also focus on such behavioral consequences as loss of control over drug use and significant disruptions in social and occupational functioning (five of seven criteria). Ultimately, understanding of the molecular, cellular, and neurocircuitry mechanisms underlying addictive disorders will provide a heuristic framework for better diagnoses.
A discussion of the physiologic mechanisms underlying addictive disease is provided in Chapters 1, 3, and 4 of this textbook as well as others. Discussion is provided therein regarding the different sites and unique mechanisms of action that drugs of abuse have in the central nervous system, underlying their acute effects and distinctive withdrawal syndromes. However, there are some common elements to addiction that are particularly relevant to the intersection of pain and addiction.
The overall hypothesis argued here is that drug addiction represents a break from homeostatic brain regulatory mechanisms that control the normal everyday emotional states of the animal, and this break in homeostasis is known as allostasis. Allostasis can be defined simply as “the ability to achieve stability through change” (88). A more complete definition is “the concept of physiology where an organism must vary all of the parameters of its internal milieu and match them appropriately to perceived and anticipated environmental demands in order to maintain stability” (88). More specifically, two components are hypothesized to comprise the allostatic break from homeostasis in addiction: underactivation of brain reward transmitter function and recruitment of brain stress (or antireward) systems. These systems (reward and stress) interact at all stages of the addiction cycle to potentiate such emotional dysregulation. Additionally, drug addiction, similar to other chronic physiologic disorders such as high blood pressure, worsens over time, is subject to significant environmental influences, and leaves a residual neuroadaptive trace that allows rapid “read-diction” even after detoxification and years of abstinence. These characteristics of drug addiction imply more than simply a homeostatic dysregulation of hedonic function but rather a dynamic break with homeostasis of these systems toward a new set point, termed allostasis as noted above.
Of relevance to issues of pain and pain treatment, the neurobiology of addiction often is characterized by two incompletely understood yet related allostatic states: tolerance and dependence. This new level of functioning that is present in addiction may be manifest not only in those brain systems in which drugs of abuse exert discrete actions (within-system neuroadaptations) but also in brain systems that oppose the actions of the drug (between-system neuroadaptations), as elaborated below (89). Importantly, the simple presence of these adaptations in the nervous system does not infer addiction. A pain patient may be tolerant to or dependent on the effects of an opioid without being addicted to it. Addiction is identified by a cluster of aberrant patterns of behavior that, while partially motivated by these physiologic changes, is evident in much broader domains; see above.
Tolerance
Ongoing use of all psychoactive drugs with addiction potential results in the development of drug tolerance, which is defined as a reduction in response to a given dose of drug after repeated administration (90,91). The neuroadaptations associated with tolerance counter the acute drug effects to maintain system-level homeostasis. A theoretical explanation for the processes underlying tolerance is offered by the opponent process theory of acquired motivation (92). Reflecting homeostatic assumptions, the theory describes how, over the course of repeated exposures to an affectively charged stimulus, a counteracting or opposing emotional response develops, which eventually accounts for habituation to the stimulus and becomes the predominant feeling state in its absence. Although opponent process theory was initially rooted in a behavioral construct, Koob et al. (93) advanced the theory by providing evidence for a neurobiologic basis to the opponent processes of tolerance and dependence in the case of addiction and specifically, for our purposes, in the case of opioid dependence. Their model predicts that in order to maintain a “normal” or homeostatic level of reward system activity, “antireward” systems are recruited to counteract drug effects, which become stronger with each exposure and extinguish more slowly than the original response (94,95). Upon abrupt drug withdrawal, the tolerance-producing processes are revealed.
Tolerance involves adaptations that occur both at the site of drug action (receptor or ion channel) (termed within system) and in systems distal to the site of drug action (96,97) (termed between system) (89). For example, tolerance to opioids is evident both at the level of opioid receptor transduction in the nucleus accumbens reward pathways and in the postsynaptic neuron at the site of this discrete drug action. Because drugs typically act at selective receptors, tolerance has been conceptualized as a functional “uncoupling” of the receptor from its effector response (opening or closing an ion channel, initiating second messenger systems); in other words, a certain proportion of receptors are rendered less functional or nonfunctional, thus making the drug less effective (97,98). An alternative form of tolerance is a between-system adaptation in which another neurotransmitter circuit becomes activated in response to sedative action in an attempt to restore homeostasis (89). Activation of an arousal system such as glutamate in the face of a sedative hypnotic drug that becomes particularly manifest during withdrawal would be an obvious example. Clinically, the resulting tolerance provides a certain amount of protection for the user, such as with the respiratory depressant effects of opioids or the anesthetic effects of ethanol.
Dependence
A related consequence of chronic drug use is dependence, which is an altered physiologic state in which an effect opposite to that of a drug becomes manifest when the drug is removed. This, of course, is intimately linked to tolerance. When a drug blood level falls below a critical point, the adaptive changes associated with tolerance predominate and become profoundly nonadaptive (99). Suddenly unopposed by drug effects, the sources of tolerance become evident as the characteristic drug-specific withdrawal syndrome (100,101) and a more generalized negative emotional state. Such negative emotional states include malaise, anxiety, dysphoria, emotional pain, loss of interest in natural rewards, and depression; are common to withdrawal to all drugs of abuse; and are particularly relevant to the interface of addiction and chronic pain.
Neurobiologic Interface of Pain and Addiction
The development of aversive or negative emotional states that constitute negative reinforcers in addiction has been linked to two distinct neurobiologic processes: loss of function in the reward systems (within-system neuroadaptations) and recruitment of brain stress or antireward systems (between-system neuroadaptations) (86,89). As addiction develops, brain stress systems that elicit release of such chemicals as corticotropin-releasing factor (CRF), norepinephrine, and dynorphin are recruited, producing aversive or stress-like states (102,103). At the same time, within the positive motivational circuits of the ventral striatum–extended amygdala, reward function is diminished. The combination of decreases in reward neurotransmitter function and potentiation of brain stress systems provides a powerful source of negative reinforcement that contributes to compulsive drug-seeking behavior and long-term dependence. Obviously, the pain itself generates a negative emotional state that can exacerbate the negative emotional state of addiction and vice versa.
Analgesic Effects of Drugs of Abuse
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


