The practice of neuropathology demands an extensive knowledge of normal central nervous system (CNS) cytology and architecture, in addition to common artifacts. During development, the CNS (from fetal life and beyond) changes dramatically, especially in terms of histology, making pathologic assessments even more challenging. A full discussion of normal CNS histology and common artifacts is beyond the scope of this text. Some of the more common neuropathologic changes and pathophysiologic processes are introduced below.
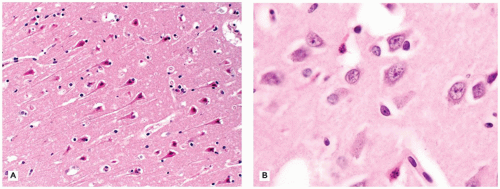
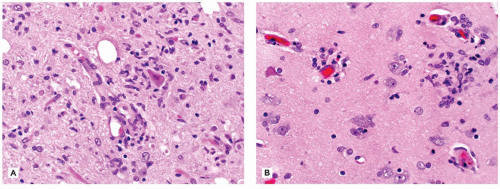
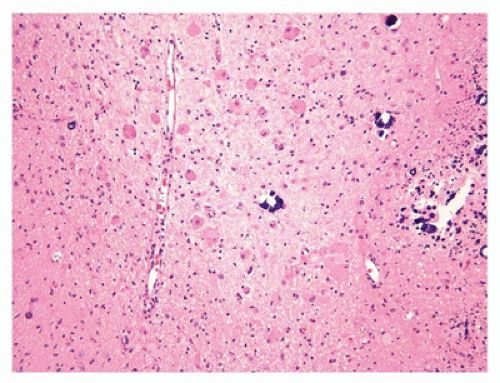
The CNS contains a variety of neurons, which vary in size from the small neocortical granular (stellate) neurons (<15 µm) to the large neocortical pyramidal neurons (measuring from 10 to 100 µm for the Betz cells of the primary motor cortex). Pyramidal neurons are often considered the morphologic prototype that bears ample lightly basophilic cytoplasm, darker clumpy Nissl substance, a large central nucleus, a prominent nucleolus (the “owl’s eye”), and coarse cytoplasmic processes. Neocortical neurons exhibit a prominent apical dendrite oriented perpendicular to the cortical surface. Neurons may display several different cytologic abnormalities, some of which are specific, but others may be nonspecific and thus requiring interpretation in the correct clinicopathologic context. Acutely necrotic neurons (i.e., “dead”) are a form of nonspecific change that contain two essential components: (a) shrunken pyknotic angular nuclei and (b) cytoplasmic eosinophilia (“red is dead”) (Figure 10-1A). Although commonly caused by ischemia (and hence often referred to as ischemic neurons), any process that causes acute neuronal death may lead to the formation of such neurons (e.g., hypoxia, hypoglycemia, carbon monoxide, epilepsy, herpes simplex virus [HSV] encephalitis). Acute neuronal death is much more difficult to appreciate in fetal brains that contain a predominance of primitive neurons (small dark nuclei with little cytoplasm); in such cases, nuclear fragmentation or karyorrhexis (at times in keeping with apoptosis) can be appreciated on high magnification (Figure 10-1B). A large variety of neuronal inclusions may be seen, including both intranuclear and intracytoplasmic types. These inclusions vary tremendously in color, size, and shape, from essentially rounded and eosinophilic/basophilic to more fibrillar (e.g., neurofibrillary tangle). These inclusions are commonly seen in viral and neurodegenerative/metabolic diseases. Abnormal vacuolization of the cytoplasm can be seen and may correlate with swelling of ultrastructural elements (e.g., mitochondria). Vacuolization that seemingly occurs within the neuropil (the meshwork of neuronal processes among which all the cell bodies of the neocortex reside) may be a result of neuronal loss, edema, or the expansion of neuronal processes (which occurs with the prion diseases or “spongiform” encephalopathies) or may be artifactual in nature. Some damaged neurons, for instance, those near infarction, may undergo mineralization (i.e., ferruginization). Damage to the axon can also lead to several cytologic alterations. Disruption of the axon may result in chromatolytic changes that include swelling of the soma, dispersion of the Nissl substance, eccentric displacement of the nucleus, and accumulation of cytoskeletal filaments. This latter event results in axonal spheroids (i.e., swellings) that can be highlighted with immunohistochemical (IHC) stains (e.g., β-amyloid precursor protein [β-APP]) (Figure 10-2). The distally transected portion of the axon degenerates and initially forms ovoids that are later taken up by macrophages, which serve to localize areas of degeneration (1). Somewhat more fusiform axonal spheroids known as “torpedoes” are commonly found in the upper cerebellar cortex and reflect Purkinje cell damage.
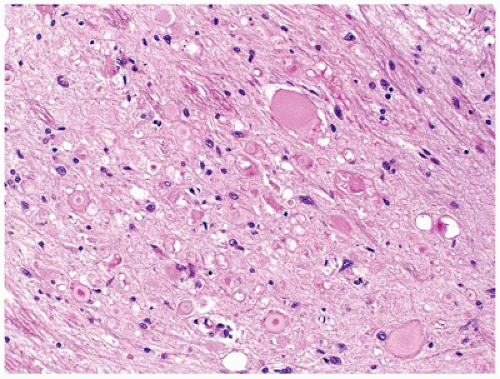
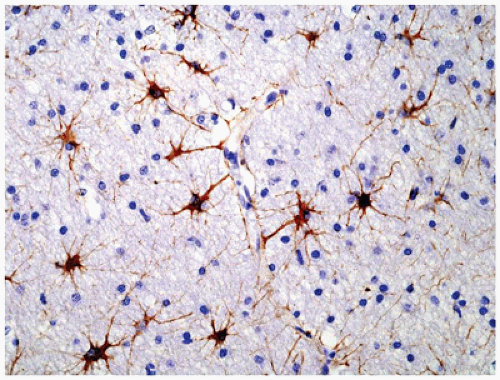
Normal astrocytes are situated throughout the gray (protoplasmic astrocytes) and white (fibrillary astrocytes) matter. For the most part, these astrocytes partake in their physiologic functions rather inconspicuously. However, in response to almost any insult, these cells undergo proliferation (i.e., hyperplasia) and enlargement (i.e., hypertrophy) termed reactive astrocytosis or gliosis, which has been equated to the “scar tissue” of the CNS (notably, the few perivascular and meningeal fibroblasts in the CNS only rarely participate to form true collagen-rich scars as seen elsewhere in the body; e.g., organizing CNS abscesses). The normally inapparent cytoplasmic processes of astrocytes (by routine H&E staining) accumulate intermediate glial fibrillary acid protein (GFAP) filaments after stimulation and thus take on a starburst-like pattern (Figure 10-3). In contrast to astrocytic neoplasms, these reactive astrocytes are typically distributed evenly throughout the parenchyma. Myelination glia (an oligodendrocyte precursor) are a notable pitfall to the determination of gliosis in the newborn; these glia also display relatively abundant eosinophilic cytoplasm, but they are a normal finding in the actively myelinating nervous system. Although their distinction from gliosis is often difficult, myelination glia often display hyperchromatic small round nuclei associated with cytoplasm that is shaped like a comet (2). In the more chronic stages of gliosis, the cytoplasmic processes of astrocytes retract and become less obvious on routine staining, although their nuclei remain in increased number and continue to mark areas of prior damage. Gliosis occurring in the cerebellar cortex in response to Purkinje cell loss is termed Bergmann gliosis, which is characterized by parallel fibrillary processes radiating through the molecular layer toward the pial surface and the accumulation of astrocytic nuclei within the Purkinje layer. The somewhat nonspecific form of gliosis that occurs immediately under the neocortical pia mater is called Chaslin gliosis, a reaction that is often attributed to previous seizure activity. Occasionally found within these areas of long-standing gliosis are the brightly eosinophilic corkscrew-shaped structures termed Rosenthal fibers (RFs) and less often eosinophilic granular bodies (EGBs); however, these structures are also seen in a variety of neoplastic (e.g., pilocytic astrocytoma [PA]) and nonneoplastic (i.e., Alexander disease) conditions (Figure 10-4). RFs have a characteristic ultrastructural appearance, manifesting as an electron-dense core surrounded by fibrillary material. An entirely nonspecific but characteristic astrocytic reaction occurs in abnormal physiologic states often associated with hyperammonemia; under these conditions, Alzheimer-type II astrocytes (of no relation to Alzheimer disease) accumulate, particularly in the basal ganglia and deep layers of the neocortex. These astrocytes, often seen in pairs, exhibit inconspicuous cytoplasm, enlarged pale nuclei, and occasionally a prominent nucleolus. Astrocytes may bear inclusions, both cytoplasmic and nuclear, although these are often difficult to appreciate on routine stains, necessitating the use of special stains and IHC for their detection.
FIGURE 10-1 • Acutely “necrotic” or dead neurons. A: Adult. B: Premature infant. Note the nuclear fragmentation (i.e., karyorrhexis) and eosinophilic cytoplasm in two shrunken subicular neurons.
FIGURE 10-2 • Axonal spheroids from a case of infantile neuroaxonal dystrophy (i.e., Seitelberger disease).
FIGURE 10-4 • Rosenthal fibers and eosinophilic granular bodies are nonspecific eosinophilic structures that are most commonly seen in the context of long-standing gliosis or within low-grade primary brain neoplasms. A: Perivascular accumulation of RFs in this case of Alexander disease. B: EGBs within the microcystic component of a pilocytic astrocytoma.
Normal oligodendroglia inconspicuously fulfill their metabolic roles (most importantly myelination) from a histologic point of view. As opposed to astrocytes, the spectrum of pathology occurring in oligodendroglia is much more restricted. By routine staining, the delicate processes of normal oligodendroglia are inconspicuous, while their hyperchromatic, round regular nuclei are sometimes surrounded by small clear haloes representing a retraction artifact of formalin fixation. As may be predicted, insults affecting oligodendroglia result in demyelination (i.e., myelin loss), which can be elucidated with myelin special stains (e.g., Luxol fast blue [LFB]). Usually, there is concomitant oligodendroglial dropout and astrocytic gliosis. Some pathologic processes cause myelin to separate between its layers, resulting in intramyelinic splitting, which manifests as vacuolar change in the white matter on light microscopy. Like astrocytes, oligodendroglia may bear abnormal nuclear (e.g., progressive multifocal leukoencephalopathy [PML]) or cytoplasmic (e.g., multiple system atrophy [MSA]) inclusions, the latter of which usually require special/IHC stains for their detection. The normally subtle cytoplasm of oligodendroglia may become more conspicuous when they suffer cytotoxic insults (e.g., ischemia).
Pathologic reactions of the ventricular lining cells, or ependyma, are generally very limited and nonspecific. These normally columnar to cuboidal cells form a simple (i.e., single layered) epithelium. With hydrocephalus (HCP) or cerebral atrophy, the epithelium stretches and becomes atrophic, or even discontinuous. Soon after acute injury, subependymal astrocytes proliferate and produce nodular excrescences that protrude into the ventricular cavity. Although previously termed granular ependymitis, this nonspecific pathologic reaction is not always related to an underlying inflammatory process; as such, the terms subventricular gliosis or ependymal granulations are preferable. If exuberant, this gliosis may entrap portions of ependyma resulting in subependymal rosettes/tubules. As with the other cellular elements of the CNS, residual ependymal cells may bear inclusions, usually of viral etiology (e.g. cytomegalovirus [CMV]).
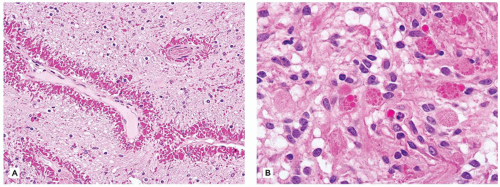
Microglial reactions are unique to the CNS. Microglia are inflammatory and antigen-presenting cells derived from bone marrow monocytes. The nuclei and cytoplasmic processes of these parenchymal cells often blend imperceptibly into the normal CNS tissue. Generally, they are of two types: (a) resident microglia are those that reside within the neuropil (and also the perivascular space) and do not undergo significant turnover with hematogenous monocytes and (b) perivascular microglia, whose population is continually renewed via hematogenous monocytes (2). Activated microglia have also been termed rod cells since, after parenchymal insult, their presence is heralded by a proliferation of small elongate naked-appearing nuclei. After CNS damage, perivascular microglia phagocytose necrotic debris and accordingly accumulate lipid material, which distends their cytoplasm yielding a foamy appearance. In contrast, resident microglia are stimulated by smaller degrees of parenchymal damage (e.g., individual neuronal necrosis), with two basic pathologic patterns seen. First, there may be a diffuse microglial activation, wherein rod cells are evenly distributed throughout the diseased tissue; some have termed this uniquely CNS reaction “neuroinflammation” (3) (Figure 10-5A). Second, and often associated with viral encephalitides, are microglial nodules, which are roughly spherical aggregates of microglia (Figure 10-5B). Microglia may also surround and digest dying neurons, a process termed neuronophagia.
FIGURE 10-5 • Activated microglia. A: Neuronophagia is seen within this diffuse microgliosis. B: In addition to some perivascular lymphocytes, a microglial nodule is seen toward the right side of the figure.
Increased Intracranial Pressure, Edema, and Hydrocephalus
Once the cranial sutures fuse early in postnatal life, the skull essentially acts as a rigid closed box, the contents of which include brain parenchyma, blood, and cerebrospinal fluid (CSF). A mature brain (approximately 1400 g) contains 75 mL each of blood and CSF (Note: a term brain weighs approximately 300 to 350 g). This CSF results in a normal intracranial pressure (ICP) of 15 mm Hg. The cerebral perfusion pressure (CPP) equals the mean arterial pressure minus the ICP. Cerebral blood flow (CBF), which for the brain is normally 50 mL/100 g/min, is calculated by dividing CPP by resistance (i.e., the vasculature). Through autoregulation, the CBF is kept constant despite changes in the systemic blood pressure. Notably, the autoregulatory capabilities of the prenatal cerebral vasculature are poor, making the brain vulnerable to fluctuations in blood pressure. When a mass-forming disease process increases the intracranial contents and elevates ICP (e.g., brain tumor), the brain compensates by expelling contents from the “closed box” so as to maintain the CBF at near normal levels. CSF leaves the cranial cavity first, and once autoregulatory mechanisms fail, blood is expelled (i.e., global ischemia), and then finally brain tissue (i.e., cerebral herniation). Several dural folds exist in the cranial cavity (e.g., cerebral falx, tentorium) and effectively serve to compartmentalize the brain. However, these extremely tough pieces of connective tissue are unyielding in the setting of increased ICP. In response to a mass lesion, brain tissue will shift or herniate from one compartment to the next, resulting in tissue damage (including contusion). Subfalcine, transtentorial (i.e., uncal), and tonsillar herniations are the most important forms, with the latter often being fatal due to compression of nearby cardiorespiratory centers in the medulla (1).
Cerebral edema is a local or generalized accumulation of fluid within the brain parenchyma that can result in increased ICP. If severe, cerebral edema may result in herniation. There are three main types of cerebral edema: (a) vasogenic, (b) cytotoxic, and (c) hydrocephalic. The blood-brain barrier (BBB) results from the specialized properties of the endothelial cells, their intercellular junctions, and a relative lack of vesicular transport (4). Breakdown of the BBB results in vasogenic cerebral edema. This type of cerebral edema is often seen in the context of CNS neoplasia and is responsive to steroid therapy. Cytotoxic cerebral edema refers to the intracellular swelling that occurs in neurons, glia, and endothelial cells. It results from failure of the ATP-dependent Na+/K+ pump and subsequent osmotic accumulation of intracellular fluids. Cytotoxic cerebral edema occurs after hypoxia, or more commonly with global ischemia due to cardiac arrest. Hydrocephalic cerebral edema is the result of transependymal CSF accumulation. Both vasogenic and hydrocephalic cerebral edema are elucidated by hyperintense signals seen on T2-weighted MRI and fluid-attenuated inversion recovery (FLAIR) sequences. Notably, these forms of cerebral edema are not mutually exclusive and often occur simultaneously. Grossly, the edematous brain exhibits congestion, with flattening of gyri and narrowing of sulci. There may be evidence of cerebral herniation (see above) manifesting as areas of necrosis and hemorrhage. Certain types of herniation routinely cause compression of large arteries and hence infarction (e.g., uncal herniation with posterior cerebral artery compression and primary occipital lobe infarction). On coronal sectioning, the ventricles are collapsed from surrounding pressure and appear slit like. By histology, the parenchyma is pale and vacuolated.
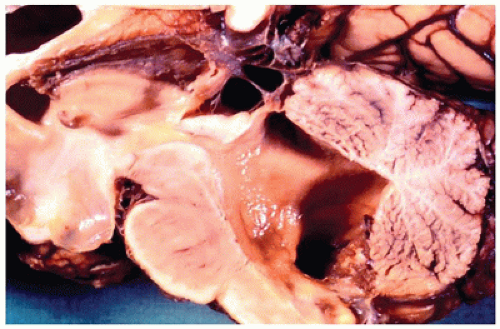
Hydrocephalus (HCP) refers to the accumulation of excess CSF and concurrent expansion of the cerebral ventricles (Figure 10-6). CSF can accumulate under normal ICP, usually in the context of cerebral atrophy; this is called HCP ex vacuo. Conventional HCP occurs under the pressure of excess CSF, usually resulting from a paucity of CSF absorption but also more uncommonly from abnormal CSF production (e.g., from a choroid plexus papilloma [CPP]). HCP due to impaired resorption can be communicating or noncommunicating, with the former resulting from a lack of arachnoid granulation-mediated CSF uptake, and the latter being caused by an obstruction in the ventricular system. Communicating HCP is often a result of meningitis or subarachnoid hemorrhage (SAH), while more rare causes include arachnoid villi aplasia or dural venous sinus obstruction. Noncommunicating HCP may be primary (i.e., congenital) or secondary (i.e., acquired). Primary causes include aqueductal stenosis (e.g., related to gliosis, atresia/forking, or an obstructing septum) and X-linked HCP (caused by mutations in the L1CAM gene on Xq28). Secondary causes include tumor, hemorrhage, or infection. Several structural CNS abnormalities may exhibit concurrent HCP (e.g., holoprosencephaly [HPE]) of unknown pathogenesis, and some suggest that the nonspecific term ventriculomegaly may be more appropriate in these cases.
FIGURE 10-6 • Hydrocephalus. There is marked dilatation proximal to and including the fourth ventricle in this sagittally sectioned autopsy of the brain, which also exhibited evidence of meningitis.
TRAUMA
Birth
Various craniospinal injuries may be mechanically incurred at birth. A number of extracranial hemorrhages may occur within the scalp whose layers can be remembered via the mnemonic “scalp” (skin, connective tissue, aponeurosis epicranialis or galea, loose connective tissue, periosteum). Hemorrhage into the subcutaneous connective tissue is called caput succedaneum. There may be subgaleal bleeding and subperiosteal hemorrhage (i.e., cephalhematoma) that often occur over the parietal bone, which may be attributable to forceps delivery. Usually, these hemorrhages resolve after a few weeks or months. Perinatal skull fractures are also frequently parietal in location and often linear in quality. Depressed skull fractures tend to be more common in children over 2 years of age. Separation of the squamous and lateral aspects of the occipital bone is called occipital osteodiastasis, and this may result in contusion of the cerebellum and posterior fossa subdural hemorrhage (SDH) (5). Epidural hemorrhage is less common than SDH and SAH. A cerebral contusion with subsequent evolution to intracerebral/intraventricular hemorrhage (ICH/IVH) is rare. However, white matter tears that are potentially hemorrhagic (i.e., gliding or internal contusions) may be seen in young infants and are thought to arise from shearing forces between the gray and white matter.
FIGURE 10-7 • Laceration and intraparenchymal hemorrhage within the spinal cord secondary to a complicated breech delivery seen here in cross sections of the spinal cord.
The spinal cord may absorb tractional or rotational forces at the time of birth. Breech and cephalic deliveries typically result in upper thoracic/low cervical and midcervical damage, respectively. Large forces can result in laceration (i.e., tearing) of the parenchyma (Figure 10-7). Petechial hemorrhages and axonal spheroids may be seen microscopically. Clinical outcome is variable; there may be acute respiratory failure and death or, in those survivors who are initially hypotonic, spasticity. The brachial plexus may be injured via tractional forces at the time of delivery. Damage to the C5-6 roots results in shoulder deficits (i.e., Erb paralysis), whereas the wrist and digits are affected with C8-T1 insult (Klumpke paralysis). Simplistically, surgical repair involves resection of the resultant traumatic neuroma with anastomosis of more normal proximal and distal nerve stumps; frozen section assessment of the degree of nerve stump viability may be requested intraoperatively.
Infancy and Childhood
Pediatric patients may suffer from both accidental and non-accidental (i.e., inflicted or abusive) injury. Motor vehicle accidents (MVAs), falls, and assaults are the leading causes of pediatric neurotrauma. The neuropathology of severe or fatal head injury in children older than 1 year is very similar to that seen in adults (1). Infants less than 1 year old suffer a different pattern of injuries, which is thought to be related to the unique and immature anatomic features seen at this age (6). These include a highly deformable skull, unfused cranial sutures, a high head-to-body ratio, an elastic spinal column with immature joints, reduced neck muscle tone, and a relatively unmyelinated brain. Accordingly, the cause of death in fatal infant craniospinal injury is often related to cerebral edema, secondary to hypoxia-ischemia. The craniocervical junction is particularly vulnerable and damage to vital brainstem cardiorespiratory centers may account for the frequent clinical presentation of apnea (6). SDH may be seen and is typically thin and bilateral (see below).
Inflicted Injury in Infants
The pathogenesis of fatally inflicted CNS injury among infants is controversial. Several terms have been used to describe the classic pattern injuries and circumstances, the most common of which is likely “shaken baby syndrome.” Many disfavor the use of this term since it implies knowledge of the mechanisms surrounding injury; hence, the term “inflicted injury” or “nonaccidental injury” is preferred. Infants suffering from inflicted injury often present clinically in a moribund state with respiratory distress or apnea. There may be lethargy, irritation, poor feeding, vomiting, and seizures. Fundoscopy may reveal retinal hemorrhages, and CT/MRI often reveals cerebral swelling and a diffuse thin layer of subarachnoid blood. In addition to inflicted mechanisms, this clinical feature may be mimicked by other etiologic entities, including MVAs, vasculopathies/coagulopathies, infection, dehydration, and metabolic abnormalities; hence, a thorough forensic-based investigation of these cases may be required (see Chapter 7).
Autopsy of an infant with inflicted injury needs to be meticulous since many findings may be subtle. Extracranial injuries may include rib and long bone fractures, which may be more conspicuous in older children. A variety of cranial fractures and hemorrhages is somewhat characteristic but nevertheless nonspecific in isolation. Skull fractures are relatively common in infants despite the inherent deformability of these bones at this age. Fractures tend to be linear and may result in dural tears and resultant CSF leaks (e.g., rhinorrhea, otorrhea). Growing fractures occur when a portion of the leptomeninges herniates through the dural defect and intercede between the two sides of a bony interruption; with time, CSF accumulates in a cyst-like space and erodes bone, thus preventing proper healing. In general, skull fractures are most often parieto-occipital. Several different types of hemorrhage may also be incurred. Epidural hemorrhage occurs between the outer surface of the dura and the adjacent skull. It is uncommon in infants, possibly because the dura is tightly adherent to the skull, and since the middle meningeal artery is more easily displaced than torn (6). SDH is common yet different than the space-occupying variety seen in older children and adults. They are thin and bilateral and are often described as “trivial.” Accordingly, infant SDHs are not thought to be due to the tearing of bridging veins, and they tend to be accompanied by retinal hemorrhages (which are somewhat nonspecific). SAH and IVHs are generally negligible. Cerebral contusions are essentially “brain bruises,” which manifest as areas of parenchymal hemorrhage and necrosis among the crests of gyri. Acutely, the hemorrhage of contusions is perivascular and oriented perpendicular to the cortical surface. Although they may be seen beneath skull fractures, contusions are generally uncommon. Cerebral edema is often the immediate cause of death and is related to global hypoxic-ischemic injury. As mentioned above, the craniocervical junction is particularly susceptible to injury, especially that which is related to stretch. Careful dissection of the cervical paraspinal muscles may reveal soft tissue hemorrhage. Spinal epidural hemorrhage may be seen but must be cautiously interpreted since this can be artifactually induced. When the craniocervical region suffers from severe hyperextension injury, pontomedullary rents (or tears) may be identified. If the injured infant survives but dies later on, signs of cerebral atrophy in keeping with hypoxic-ischemic encephalopathy (HIE) may be seen.
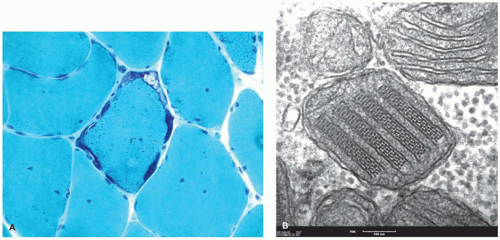
Although diffuse axonal injury (DAI) was previously purported to be the underlying neuropathology in infant-inflicted injury, it is only identified in rare cases As opposed to DAI, the axonal damage in infant-inflicted injury is localized often to the lower pons and upper medulla and particularly to descending corticospinal tracts (7). In addition, axonal damage may be apparent in the cervical and other spinal roots (8). Axonal spheroids can be seen on routine stains about 24 hours after injury, but IHC staining for β-APP can highlight microscopic axonal damage 2 hours or less after injury (9). The lysosomal marker, CD68, is often used to label microglia and may be used to highlight the acute and more remote cellular reactions to axonal damage. HIE is frequent and is seen more diffusely throughout the brain; usually, HIE is the most prominent finding in cases of infant-inflicted injury. Early features include acute neuronal necrosis, parenchymal vacuolation, microglial activation, and myelin pallor, whereas in chronic stages, parenchymal rarefaction predominates with neuronal loss and gliosis.
SUDDEN INFANT DEATH SYNDROME
Sudden infant death syndrome (SIDS) is a complex multifactorial disorder. SIDS is generally defined as the sudden unexpected death of an infant (<1 year in age) that often occurs during sleep and for which no cause of death is identified despite thorough history, death scene investigation, and autopsy. Numerous factors are associated with SIDS, but the role to which each impacts this disorder remains elusive. Many of these factors are hypothesized in terms of their effect on the infant’s innate autonomic physiologic response to normal homeostatic stressors (e.g., apnea, hypercarbia/blood pH, hypotension). These infant responses are largely mediated by brainstem nuclei. Maternal factors increasing the risk of SIDS include low socioeconomic status, low maternal education, cigarette smoking, and alcohol consumption. These maternal factors may play a role in predisposing the embryo/infant to hypoxic-ischemic damage (e.g., periventricular leukomalacia [PVL]) or brainstem abnormalities, which can be seen pathologically in a subset of SIDS cases (10); notably, the histopathologic changes seen in SIDS cases are often inconspicuous. Cerebellar and more so medullary abnormalities have been hypothesized. Derivatives of the rhombic lip (e.g., external granule layer [EGL] of the cerebellum, the inferior olive, and the arcuate nucleus) have also been implicated in SIDS. More recently, focus has been directed toward possible abnormalities in the medullary serotonergic (5HT) nuclei, as championed by Hannah Kinney and colleagues (11,12). These nuclei have been suggested to normally modulate and integrate autonomic, respiratory, and somatomotor responses to homeostatic stressors; in particular, lowered 5HT receptor binding has been demonstrated in the arcuate nucleus (13). Susceptibility genes have been proposed, as evidenced by the detection of polymorphisms in the promoter regions/coding sequences; candidate genes include the 5HT transporter, IL-10, and heat shock protein 60 (6). Rare SIDS-like cases have even been associated with cardiac sodium channel mutations (causing arrhythmias) and inborn errors of metabolism (especially medium chain acetyl-CoA dehydrogenase deficiency). In addition to its association with sleep, death in SIDS is often associated with the prone position. Integration and synthesis of these factors have led to the triple-risk model of SIDS (14). This model proposes that there are three key factors leading to infant death when present simultaneously: (a) a vulnerable infant (i.e., those with pathophysiologic abnormalities), (b) a critical period of development (the peak incidence of SIDS cases occurs at 2 to 4 months and may be related to brain maturation), and (c) an exogenous stressor (e.g., prone sleeping). As researchers continue to unravel the mysteries of this disorder, management and minimization of these recognizable risk factors are the best means of preventing this devastating syndrome (see Chapter 7).
STRUCTURAL MALFORMATIONS OF THE CNS
Neural Tube Defects, Axial Mesodermal Defects, and Tail Bud Defects
Classic embryology describes three primitive germ layers: endoderm, mesoderm, and ectoderm. Near 16 days post-ovulation, the mesodermally derived notochord induces the development of CNS tissue from the overlying ectoderm; the signaling molecule sonic hedgehog (Shh) is important to this process. This newly formed neuroectoderm first thickens into the neural plate. A longitudinal neural groove then develops, and subsequently at 18 to 20 days postovulation, neural folds arise from the lateral aspects of the plate. The neural crest (the forerunner of the spinal, cranial nerve and autonomic ganglia, leptomeninges, Schwann cells, melanocytes, and other tissues) originates from the apices of these folds, which eventually meet at distinct closure sites in the midline to form the neural tube. In humans, two initial closure sites are well recognized, one (site 1) at the cervical-occipital boundary (on day 22 postfertilization) and a second (site 2) at the extreme rostral end of the neural plate. A third closure site at the forebrain-midbrain boundary may also exist. Fusion of the neural fold proceeds bidirectionally from site 1 and caudally from site 2. Fusion of the cranial portion of the neural tube is completed at the anterior neuropore (24 days postfertilization), which subsequently develops into the lamina terminalis. The caudal aspect of the neural tube finishes closure at the posterior neuropore (28 days postfertilization). In general, the process of neural tube formation is called neurulation and is divided into two aspects: (a) primary neurulation describes the fusion of neural folds, which form the rostral aspects of the CNS, and (b) secondary neurulation describes the formation of the caudal-most neural tube (i.e., lumbosacral spinal cord) that occurs through canalization of a solid mass of cells. Primary and secondary neurulated tissues eventually join to form the complete neural tube. After neural tube formation, the axial skeleton begins its development and eventually encases the maturing CNS. The skull has a dual origin: the cranial vault and occiput develop from axial mesoderm (endochondral bone formation), while the skull base and facial bones arise from the cranial neural crest (membranous bone). The vertebrae also arise from the axial mesoderm.
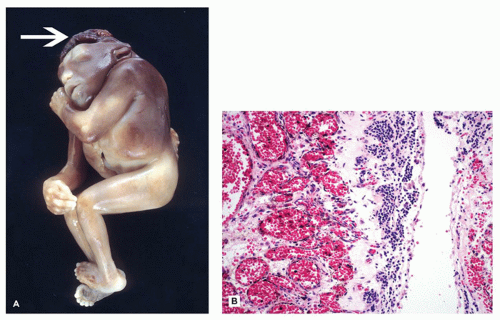
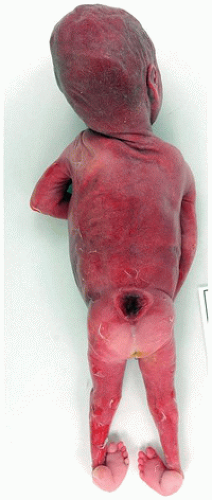
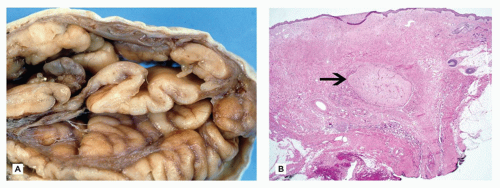
Neural Tube Defects
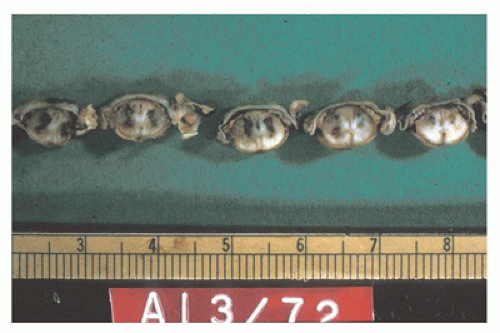
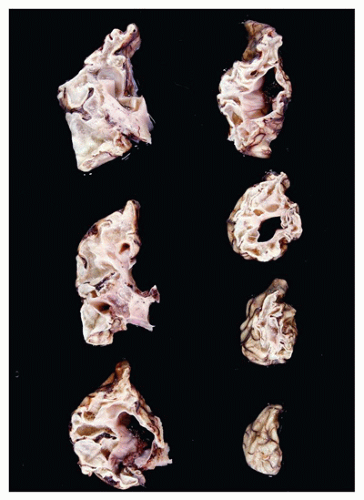
Neural tube defects (NTDs) are the result of defective neural tube closure during the 3rd to 4th week of gestational age (GA). The true incidence of NTD is difficult to determine since severe forms lead to spontaneous abortion. Birth prevalence has been estimated to be between 3/1000 and 7/1000 depending on several factors, including geography (e.g., high prevalence in Northern Ireland) (15). There is a spectrum of CNS involvement, from widespread (e.g., complete craniorachischisis) to more focal (e.g., lumbosacral myelomeningocele). Patients with focal spinal forms may survive with motor and sensory deficits below the level of nonclosure, which include rectal and urinary sphincter involvement (e.g., incontinence, urinary tract infections). Additional problems may include HCP, Chiari II malformation, and kyphosis. Although the use of folic acid supplements has reduced the frequency of NTDs, the therapeutic mechanism remains unclear. Craniorachischisis is the most severe form of NTD wherein there is complete failure of neural tube closure such that the brain and spinal cord are exposed to the amniotic fluid. There may be some forebrain development rostrally, but neural tube closure is usually deficient distal to the midbrain. In anencephaly, which is now usually encountered near midgestation after termination of pregnancy, the defective neural tube is generally limited to the cranial and cervical regions (Figure 10-8A). The skull vault is absent, the skull base is malformed (thick and flat anomalous sphenoid bones), and the orbits are shallow. The majority of the brain (minus portions of the anterior pituitary, cranial nerves, medulla, and some cerebellar folia) is replaced by the area cerebrovasculosa. This CSF-filled cystic angiomatous area contains neuroglial-derived tissues (including ependyma, neurons, neuroblasts, and choroid plexus) and numerous thin-walled, closely packed, and congested blood vessels. Contrary to most descriptions that describe disorganization, the neuroglial and vascular components of the area cerebrovasculosa are usually quite organized across cases and produce a laminar structure; the neuroglial tissue, which often has a nodular substructure, forms a layer that is usually more superficially situated than the vascular layer (Figure 10-8B). Occasionally, a third layer composed of macrophages is seen actively breaking down the primitive neural tissue. Atrophic, keratinizing, squamous epithelium that is continuous with normal skin covers the defect. Due to the deficiency of cerebral tissue, there is a paucity of descending spinal cord tracts. Overlying spinal leptomeninges are vascular and may contain glioneuronal heterotopias (6). Associated non-CNS abnormalities include hypoplastic adrenals and lungs, plus an enlarged thymus (16). Myelomeningoceles can occur throughout the spinal cord, but those affecting the lumbosacral region are the most frequent (Figure 10-9). An association with the Chiari II malformation is frequently appreciated in those cases with lumbosacral myelomeningoceles, prenatal surgical closure of which has been shown to mitigate clinical sequelae (17). The spinal cord may be “closed” (i.e., no NTD per se) or “open” posteriorly as a flattened lesion. Closed lesions are cystic and covered by a delicate membrane/skin that contains a hydromyelic cord. Open lesions contain a vascularized mass of disorganized neuroglial tissue called the area medullovasculosa that is covered by atrophic cutaneous tissue. In either case, the spinal cord and meninges herniate through an associated vertebral defect. There may be additional spinal cord abnormalities above the NTD (e.g., hydromyelia, syringomyelia, and diplomyelia).
FIGURE 10-8 • Anencephaly. A: Because of the absent calvarium (or “skull cap”), malformed vascularized neuroglial tissue (i.e., area cerebrovasculosa) can be directly visualized (see arrow). B: Microscopically, the area cerebrovasculosa usually displays a very organized layered architecture, with a superficial primitive neural layer and a subjacent vascular layer (H&E, 200×).
FIGURE 10-9 • Lumbosacral myelomeningocele, 23 weeks gestational age. This NTD was associated with Chiari II type changes in the brain. In passing, note the clubbed foot deformity.
Herniation Through Axial Mesodermal Defects
Portions of CNS tissue (with proper neural tube closure) may herniate through axial mesodermal (i.e., bony) defects. These include encephaloceles and meningoceles. Encephaloceles may be anteriorly located (frontoethmoidal cases are common in Southeast Asia), but occipital cases are the most frequent (Figure 10-10). Occipital encephaloceles can involve the foramen magnum and include portions of the cerebellum, brainstem, and occipital lobe (e.g., Chiari III malformation). Meckel-Gruber syndrome is a lethal autosomal recessive disorder that is characterized by the triad of CNS malformations (especially occipital encephalocele), cystic dysplasia of the kidneys, and ductal plate malformations of the liver (18). It can be detected by ultrasound prior to 14 weeks’ gestation. There is genetic linkage to three loci: 17q21-24 (MKS1), 11q13 (MKS2), and 8q24 (MKS3). Meningoceles are typically lumbosacral in location. All three layers of meninges herniate through the bony vertebral defect, while the spinal cord remains in a normal position. Accompanying spinal cord defects may be seen and include hydromyelia, syringomyelia, diastematomyelia, and cord tethering.
Tail Bud Defects
Tail bud defects are thought to involve abnormalities of secondary neurulation. Cord abnormalities are lumbosacral and include hydromyelia (dilatation of the central canal), diastematomyelia (splitting of the cord into hemisections, often due to a bony spur), diplomyelia (duplication of the spinal cord), and cord tethering. The tethered cord syndrome per se involves lower limb motor and sensory deficits, pain, and neuropathic bladder, all of which presumably result from traction on distal cord elements. There may be a thickened filum terminale, low or dilated conus medullaris, spinal lipoma, or other abnormalities in the lumbosacral cord or sacral region in general (6). Detethering frequently leads to clinical improvement. Surgical specimens often reveal an increase in dense collagenous tissue and foci of fat cells, the latter of which is not a normal constituent of the filum terminale (19).
FIGURE 10-10 • Encephalocele. A: Atrophic cutaneous tissue overlies brain parenchyma in this surgical specimen. (Image courtesy of Dr. Beth Levy, St. Louis University.) B: Histologically, neuroglial tissue (arrow) is embedded within the deep subcutaneous connective tissue from a more subtly involved example.
Disorders of Forebrain Development
The early development of the forebrain and midline structures, as it pertains to the neuropathology of structural malformations, is described in greater elsewhere (1,20). In brief, three primary brain vesicles are present by the 4th week of GA: (a) prosencephalon (forebrain), (b) mesencephalon (midbrain), and (c) rhombencephalon (hindbrain). Forebrain induction is thought to be governed by the prechordal plate, the ventralizing molecule Shh, and the dorsalizing molecule bone morphogenic protein 7. By constraining growth in the ventral midline of the forebrain primordium, the relatively rapid dorsolateral growth leads to the formation of paired telencephalic secondary vesicles by the 6th week GA. There are five secondary brain vesicles: (a) the telencephalon (cerebral hemispheres and basal ganglia) and (b) diencephalon (thalamic substructures), both arising from the prosencephalon; (c) the mesencephalon; (d) the metencephalon (pons and cerebellum) and (e) myelencephalon (medulla), both arising from the rhombencephalon. Shh also induces the optic primordium to divide and grow out from the diencephalon at 4 to 5 weeks GA. The paired olfactory vesicles are induced by the olfactory placodes and their ingrowing olfactory nerves at 6 weeks GA. The anterior commissure begins its development at 10 weeks GA, arising from or adjacent to the lamina terminalis. At this same time, the fornices and hippocampal primordia arise nearby and grow in a reverse C-shaped manner en route to their destination in the temporal lobe. The corpus callosum arises at 12 weeks GA from the massa commissuralis, which is slightly rostral and superior to the anterior commissure. It grows in a rostrocaudal manner and in doing so results in the formation of the septum pellucidum at 20 weeks GA.
Holoprosencephaly and Agenesis of the Corpus Callosum
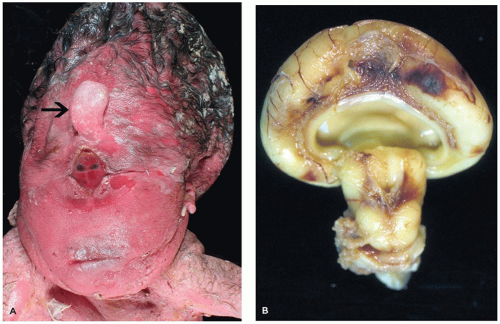
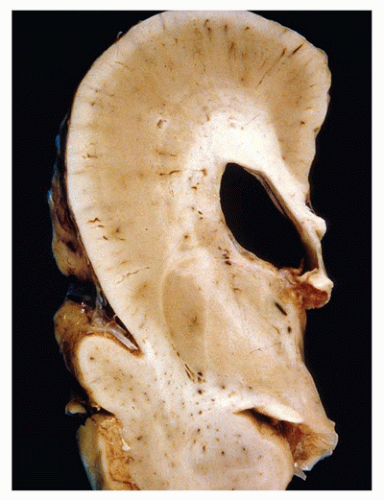
HPE is a disorder of induction and patterning of the rostral neural tube occurring at 4 to 5 weeks GA. This may be the result of a faulty prechordal plate (21). The majority of cases are sporadic, and the incidence is approximately 5/100,000 to 9/100,000 live births. Risk factors for HPE include maternal diabetes and possibly alcohol consumption. Chromosomal abnormalities are commonly seen, the most frequent of which is trisomy 13 (Patau syndrome). Molecular genetic investigations have revealed several genes that are potentially mutated in HPE, including SIX3 (HPE2), SHH (HPE3), TGIF (HPE4), ZIC2 (HPE5), and PTCH (HPE7). Mutation of the sonic hedgehog gene (SHH) is most commonly seen, a finding that is especially intriguing in light of its normal role in neuroectodermal induction. HPE may be seen in the context of a well-recognized syndrome, such as Smith-Lemli-Opitz syndrome (due to a defect in cholesterol biosynthesis). Notably, the Shh molecule must undergo autoproteolytic cleavage for proper functioning, which in turn is dependent on cholesterol attachment to its carboxy-terminus. HPE has a variable clinical picture; severe forms result in death early in life or midgestational termination, while more mild forms allow survival into adulthood. Craniofacial and ocular abnormalities that can be present have been hypothesized to be a result of defective mesencephalic neural crest (22) (Figure 10-11A). Microcephaly (i.e., small head) is common; brains are correspondingly micrencephalic (i.e., low weight) and often less than 100 g at term. There may be hypotonia, seizures, developmental delay, and cognitive impairment. Hypofunctioning of the pituitary gland results in pan-endocrinopathies.
FIGURE 10-11 • Alobar holoprosencephaly. A: Examination of the face reveals a proboscis (arrow), which is superior to a single orbit bearing two fused globes. B: Superior and caudal views of the brain reveal the horseshoe-shaped holosphere containing a single ventricle that opens posterodorsally. (Images courtesy of Dr. Robert Schmidt, Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO.)
There are three main clinicopathologic categories of HPE that together likely represent a spectrum of disease severity. Lobar, semilobar, and alobar forms correspond to increasingly severe structural and clinical diseases, which is generally defined by the extent of the midline longitudinal fissure. In less severe forms, the fissure can be seen “cleaving” the cerebrum into two cerebral hemispheres more caudally. In alobar HPE, the most commonly encountered form on the pediatric neuropathology service, the longitudinal fissure is absent and results in a single cerebral mass or “holosphere.” The Sylvian fissure, the gyrus rectus, and the olfactory structures are also absent. This anomalous gyral pattern prohibits the delineation of cerebral lobes. The holosphere is horseshoe shaped and contains a single ventricle that opens posterodorsally (Figure 10-11B). The opening of this ventricle is covered by a delicate membranous roof that attaches to the tentorium; this membrane may balloon to form a dorsal cyst. The lateral aspects of this membrane are bounded by a single, arch-shaped hippocampus. The floor of the single ventricle is formed by the fused deep gray nuclei. Although the corpus callosum is absent, there is no bundle of Probst (see below). The anterior commissure and the septum pellucidum are also absent. Both the brain stem and the cerebellum are grossly normal (with the exception of hypoplastic corticospinal tracts). The skull base is malformed. The anterior aspects of the circle of Willis are anomalous, and both the anterior and middle cerebral arteries are replaced by a disorganized collection of vessels called the rete mirabile. Microscopically, the cortical gray matter is occasionally dysplastic (1,16,23). In affected cases, it is excessively thick and dyslaminated and may demonstrate a progressively abnormal lateromedial gradient of architectural disturbance (22). The sparsely cellular external layer is segmented and arranged into irregular clusters, which may form thick cords of neurons that can traverse the entire pallium (i.e., developing cortical gray matter). There may be acellular deep zones or “glomeruli.” The deeper neocortical neurons are often maloriented. Norman et al. suggest that the abnormal cortex seen in the context of HPE is unique to this disorder (16). The leptomeninges of the holosphere can be laden with glioneuronal rests and form a superficial “crust” over the brain, reminiscent of leptomeningeal neuroglial heterotopia. The architecture of the hippocampi, deep gray nuclei, and cerebellum is also often abnormal, with the latter exhibiting dysplasia, heterotopia, and an association with trisomy 13. The least severe lobar form of HPE, despite its resemblance to a normal brain, still contains the cerebral cortex that is continuous across the midline (at the frontal pole, in the orbital region or above the corpus callosum causing cingulosynapsis) (Figure 10-12). Portions of the olfactory structures and posterior corpus callosum may be present. Semilobar HPE is intermediate in appearance. In the recently described middle hemispheric variant of HPE, portions of the deep gray nuclei and frontoparietal lobe are fused across the midline, with relative rostral, caudal, and ventral brain sparing.
FIGURE 10-12 • Cingulosynapsis. This 24- to 25-week gestational aged fetus with semilobar holoprosencephaly displays midline fusion of the rostral most aspect of the cerebral hemispheres (top right). In addition, grey matter (arrows) located in a region consistent with the cingulate gyrus is seen abnormally crossing the midline (i.e., cingulosynapsis); note that the corpus callosum, which is normally well demarcated from the overriding cingulate gyrus, is ill defined.
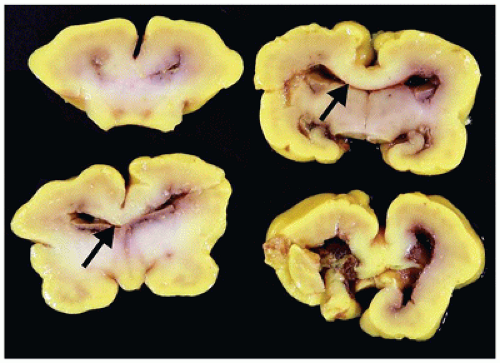
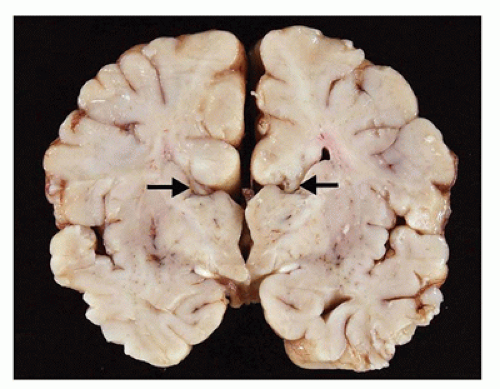
Agenesis of the corpus callosum (ACC) may be isolated or seen in combination with other CNS abnormalities. These associations (e.g., a neuronal migration disorder [NMD]) make it difficult to assess the clinical impact of ACC per se. However, isolated ACC is most often asymptomatic and found incidentally on imaging. Potential signs and symptoms may include seizures, cognitive impairment, subtle perceptual deficits, or a disconnection-like syndrome. ACC may be associated with a well-recognized syndrome (e.g., Aicardi) or an inborn error of metabolism (e.g., nonketotic hyperglycinemia). Pathologically, the characteristic findings of ACC are seen grossly on coronal sectioning of the brain (24). Agenesis may be complete or partial, with latter forms being found more caudally (i.e., splenium) in keeping with the rostrocaudal embryologic development of the corpus callosum. Laterally situated and longitudinally orientated bundles of white matter are usually identified immediately superior to the lateral ventricles; these are called the bundles of Probst and are thought to represent misdirected callosal fibers (Figure 10-13). The normal dorsolateral angles of the lateral ventricles take on an abnormal superior orientation (i.e., “batwing ventricles”). The distended membranous roof of the 3rd ventricle displaces the fornices and leaves of the septum pellucidum laterally. The cingulate gyrus is replaced by several short radiating gyri and the anterior commissure may also be absent; these features are best appreciated on medial inspection of the brain prior to coronal sectioning of the cerebral hemispheres. Other structural abnormalities that may accompany ACC include HCP (in this context, often occurring in the posterior cerebral hemispheres and termed colpocephaly), olfactory hypoplasia, and NMDs (see below). A subset of ACC may be the result of mechanically impeding mass lesion (e.g., lipoma), but the pathogenesis of most other cases is unclear. Some have speculated that abnormalities in the “glial sling,” which normally guides commissural fibers across the midline, may be a potential cause of ACC (25).
FIGURE 10-13 • Agenesis of the corpus callosum. A 2-month-old infant male. History of sudden unexpected death and cosleeping. Coronal sections of the brain do not reveal a normal corpus callosum; in its absence, dorsolaterally directed “bundles of Probst” (arrows) are noted.
Other disorders of forebrain induction include olfactory aplasia, atelencephaly, aprosencephaly, and abnormalities involving the septum pellucidum (1,2). Just as the development of the cingulate gyrus and that of the corpus callosum are linked, so are those of the olfactory bulbs and the gyrus rectus; true olfactory aplasia is usually accompanied by the absence of the gyrus rectus.
Cell Migration and Specification Disorders
The embryology of early neocortical development is reviewed in greater detail by Golden and others (6,20). The wall of the early neural tube is composed of a pseudostratified neuroepithelial layer. By 4 weeks GA, the outer preplate zone has emerged from the inner ventricular zone of neuroepithelium. The preplate is composed of two layers: an outer layer of Cajal-Retzius cells (CRCs) (i.e., neurons) and an inner layer of subplate neurons. These transient subplate neurons play an important role in the early organization of neocortical connectivity. CRCs secrete reelin, an important extracellular matrix protein that assists migrating neuroblasts in finding their correct neocortical laminar destination. By 6 weeks GA, radial glia processes have essentially spanned the cortical mantle and aligned themselves perpendicular to the brain surface. These radial glia processes serve as physical guides for the migrating neuroblasts, which will form the cortical plate and eventual neocortex. The process of radial migration is complex and involves numerous molecules, including neuregulin, ErbB4, cell adhesion molecules, astrotactin, extracellular matrix molecules, and their receptors (6,20). A two-part physical barrier to overmigration lies in the marginal zone (i.e., laminae I) of the pallium and is composed of (a) the glia limitans, which is formed by the expanded end feet of the radial glia and the basal lamina of pial blood vessels, and (b) the horizontal processes and synapses of CRCs. Neuroblasts leave the ventricular zone to populate the cortical plate, “split the preplate,” and form the future neocortical laminae in an inside-to-outside sequence, with the deepest layers forming prior to more superficial layers (i.e., laminae VI prior to V). These neuroblasts migrate in waves between 6 and 20 weeks GA. Despite this migration, histologic evidence of lamination in the neocortex is essentially absent at midgestation (save for the molecular layer) and the neocortex forms a very cell-dense and well-delimited band. Over the subsequent 10 weeks, neocortical lamination gradually emerges, and the 6-layer appearance is often appreciated by 30 weeks’ gestation. In addition to radial migration, there is also tangential migration to the developing neocortex. In tangential migration, migrating neuroblasts are guided by neuronal processes to their destination; this form of migration is utilized by the future inhibitory interneurons of the neocortex that arise from the medial and lateral ganglionic eminences (i.e., the germinal matrix) and travel along axonal processes that run parallel to the cortical surface. Initially, the primitive neocortex is overpopulated by neurons, and their numbers are normally culled by apoptosis, the latter of which is usually difficult to identify with any significant frequency in fetal autopsy brains. The remaining neurons terminally differentiate and establish the connectivity pattern indicative of the developed neocortex.
Lissencephaly, Types I and II
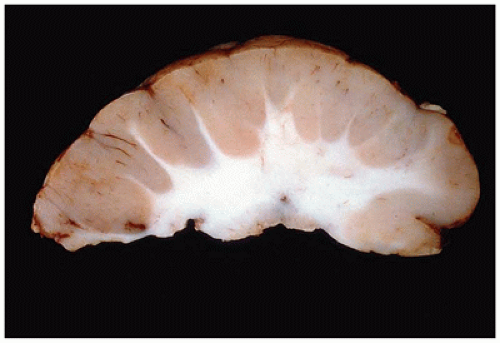
Lissencephaly type I (i.e., classical type) is a diffuse and abnormally “smooth” (i.e., agyric) cerebral surface, whereas pachygyria represents a more focal agyric abnormality among more normally gyrated cortex. Lissencephaly type I is caused by disrupted neocortical cell migration. A number of additional CNS malformations may be associated with lissencephaly type I, and imaging that reveals such features can guide genetic testing. Four main genes have been linked to lissencephaly type I and some appear associated with distinct histopathology (6). LIS1 (17p13.3) encodes the LIS1 protein [aka platelet-activating factor acetyl hydrolase 1 subunit β1 (PAFAH1β1)] that is involved in a complex cellular cascade, which influences dynein (and hence cell movement) and possibly cell proliferation. LIS1 is associated with Miller-Dieker syndrome. XLIS (or DCX; Xq22.3-23) encodes doublecortin, which is a microtubule-associated protein; while affected males have lissencephaly type I, females exhibit subcortical band heterotopia (SBH; see “Cerebral Heterotopia” below). The reelin protein (encoded by the RELN gene; 7q22) is the extracellular ligand that influences intracellular downstream targets (e.g., LIS1 protein). Clinically, RELN mutation causes lissencephaly type I associated with cerebellar malformations. Finally, ARX (Xp21) encodes a transcription factor important in CNS/PNS development; mutations result in lissencephaly type I and ambiguous genitalia. The general clinical features of lissencephaly type I include developmental delay, cognitive impairment, seizures (including infantile spasms), and microcephaly.
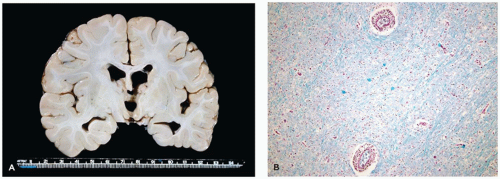
Pathologically, lissencephaly type I is characterized by thickened neocortical gray matter and a paucity of white matter (Figure 10-14). The agyric cortex may be preferentially seen more rostrally (XLIS or RELN mutation) or caudally (LIS1 mutation). Foci of pachygyria tend to have an ill-defined border with more normal brain. Heterotopic gray matter may be seen in the periventricular and deep white matter, and if associated with an XLIS mutation, a subcortical band of gray matter may be seen. There may be HCP. The cerebellum is typically hypoplastic in cases associated with RELN mutations. The inferior olives may be dysplastic, and the corticospinal tracts may be abnormal. Classically, the histology of lissencephaly type I is described as a malformed four-layer cortex: (a) the outer first layer (i.e., molecular layer) is fairly normal and contains CRCs, (b) the second layer contains large maloriented pyramidal neurons, (c) the cell poor third layer may be myelinated in older children, and (d) the fourth layer is thick and contains disorganized small to medium pyramidal and granular neurons. This classic four-layer pattern corresponds to the LIS1 mutation and is more prominent in posterior cerebral aspects. Recent investigations have suggested additional histologic variants, including four-layer anteriorly predominant (DCX mutation), three-layer (ARX mutation), and two-layer forms (26).
FIGURE 10-14 • Lissencephaly type I. The markedly thickened cerebral gray matter displays an absence of gyration. There is a concomitant paucity of cerebral white matter (coronal section). (Image courtesy of Dr. Beth Levy, St. Louis University.)
The cell migratory defect in lissencephaly type II (i.e., cobblestone type) appears to be one of overmigration. A defective glia limitans allows radial glial processes to extend beyond the normal limits of the neocortex, facilitating the excessive migration of neuroglial precursors. Lissencephaly type II shares a thickened neocortical gray ribbon and at times an agyric surface with lissencephaly type I. However, the five main autosomal recessive syndromes associated with lissencephaly type II exhibit a characteristic triad of cerebral, ocular, and muscle diseases that are not seen in type I disease. These syndromes include Walker-Warburg syndrome, Fukuyama congenital muscular dystrophy (FCMD), muscle-eye-brain disease, congenital muscular dystrophy type 1D (MDC-1D), and MDC-1C, a disorder associated with fukutin-related protein (FKRP) mutations. FCMD is the most common (incidence of 3/100,000/year) and characteristically occurs in Japan. The abnormalities in these syndromes (and hence lissencephaly type II) are thought to be due to defective glycosylation, in particular O-mannosylation. Glycosylation is a common posttranslational protein modification and, in general, is important to normal development. O-glycosylation of α-dystroglycan appears particularly important to the etiology of these conditions (27). Pathologically, these micrencephalic brains have a lissencephalic cortex that may have a “bumpy” quality (i.e., cobblestone). The gray-white junction tends to be distinct below the thickened gray matter. The white matter is deficient and there may be HCP. The brain stem is small, in part due to hypoplastic corticospinal tracts. The cerebellum in these cases is characteristically small, especially in the vermal region, and cases of Walker-Warburg syndrome may exhibit features of a Dandy-Walker malformation (DWM) and an occipital encephalocele. Microscopically, the cortex is very disorganized and unlaminated. Superficial aspects tend to be more abnormal and may resemble polymicrogyria. The gray-white junction may exhibit a nodular appearance. The deep and superficial areas are separated by large internalized and hyalinized blood vessels that likely represent the original, and overrun, leptomeningeal vasculature. Less severely affected areas may contain a leptomeningeal “crust” of glioneuronal heterotopia. The cerebellum is disorganized, and although the internal granular and Purkinje neurons retain their somewhat normal relations, the normal architecture is disrupted. Bands of white matter are seen over the cerebellar surface. The overall appearance of the cerebellum may also resemble polymicrogyria.
Polymicrogyria
Polymicrogyria is a cortical malformation where the neocortical gray matter ribbon may seem grossly thickened but microscopically is actually thin, excessively folded, and fused. Intrinsic and acquired origins for this lesion have been proposed (6). The risk factors for polymicrogyria include (a) intrauterine infection (e.g., “TORCH”), (b) intrauterine ischemia, (c) metabolic diseases (e.g., Zellweger syndrome), and (d) a family history. Polymicrogyria may also be associated with well-recognized syndromes. Karyotypic abnormalities have been noted (e.g., -1p36, -22q11), but with the exception of FGFR3 mutations in thanatophoric dwarfism, specific mutational information is limited (28). Clinically, localized polymicrogyria may be asymptomatic, but more often, it is associated with developmental delay, psychomotor retardation, spastic diplegia, pseudobulbar palsy, and epilepsy. MRI highlights this abnormal cortex and may reveal additional structural abnormalities (e.g., decreased white matter or other white matter changes, calcification, schizencephaly, porencephaly). Grossly, the cerebral surface in polymicrogyria is irregular and bumpy. Coronal sections reveal thickened neocortical gray matter composed of serpiginous, heaped-up thin layers. Polymicrogyria may be widespread and symmetric or focal and asymmetric. Cingulate and striate cortices are often spared. Polymicrogyria may be seen in the relatively spared temporal lobe of hydranencephaly or adjacent to porencephalic defects. Microscopically, the cortex is composed of numerous attenuated, excessively folded, and fused layers (Figure 10-15). Fusion of adjacent molecular layers results in a branching pattern of paucicellular tissue, which often bears a central blood vessel. The cortex is usually unlayered but may be four layered (similar to lissencephaly type I). However, Judkins et al. suggested that categorization of polymicrogyria by lamina is artificial since the neocortical laminae are normally formed but neuronally depleted; as such, this group suggested that polymicrogyria is not a cell migratory disorder, but rather a postmigrational malformation of cortical development (29). In passing, leptomenin-geal glioneuronal and nodular heterotopias may also be seen alongside polymicrogyria.
FIGURE 10-15 • Polymicrogyria. Transverse sectioning of this surgical brain specimen reveals abnormal undulation of the cortical ribbon.
Cerebral Heterotopia
Cerebral heterotopia refers to malformative lesions wherein groups of cytologically normal brain cells (i.e., neurons and glia) do not reach their neocortical destination. Three main categories are discussed here: leptomeningeal heterotopia (LH), periventricular heterotopia (PH), and SBH. LH and PH are often associated with other CNS malformations, while SBH is usually seen in isolation. LH is likely the most common of these three forms and is usually focal. Genetic and epigenetic risk factors are associated with each form of cerebral heterotopia. Genetic syndromes linked to LH include trisomy 13, HPE, and lissencephaly type II. The genetics of PH are complex, but one X-linked form involves mutations of FLNA (Xq28) (30). SBH is usually due to mutations in the XLIS gene (i.e., doublecortin, DCX) (see above). Epigenetic risk factors likely represent the most common mechanisms underlying LH/PH and include HIE, PVL, and germinal matrix/subpial hemorrhage. Damage to the glia limitans and radial glia likely underlies the pathogenesis of LH and PH, respectively. It is difficult to assess the clinical impact of these heterotopias since LH and PH are frequently associated with other CNS malformations. However, cognitive impairment and seizures often accompany all three forms of cerebral heterotopia. Grossly, LH is often inapparent unless seen in the context of lissencephaly type II. SBH appears as a band of gray matter (outside of the intragyral white matter) that is flanked on either side by white matter. On close inspection, the gray matter of the SBH may be broken up into nodules, which are split by white matter bundles. PH may also be confluent (i.e., band-like) or nodular, the latter being more common. Like LH, PH is associated with abnormal adjacent neocortex (cortical dysplasia with LH and polymicrogyria with PH). Microscopically, all three forms of cerebral heterotopia appear similar. Pyramidal and granular neurons are associated with glia and other normal neocortical elements, but there is no lamination and the neurons are maloriented. In LH, there is usually some connection to the underlying cortex.
“Malformations of Cortical Development,” Including Focal Cortical Dysplasia
This group of epileptogenic CNS malformations regrettably has been plagued by a plethora of confusing terminology. Malformations of cortical development (MCD) are now the preferred umbrella term that encompasses several entities including the NMDs (see above), focal cortical dysplasia (FCD), and microdysgenesis. Previously, the term “cortical dysplasia” was often used nonspecifically to describe many different abnormal cortical histologies. “Microdysgenesis” has similarly been utilized in the past to describe milder abnormalities in cortical architecture (e.g., excess white matter neurons, excess perivascular oligodendroglia in the white matter, “glioneuronal hamartia,” abnormal neuronal clustering, dyslamination, cortical columnarization) (31); however, the term mild MCD is now favored over microdysgenesis (see below). At the time of publication of the previous edition of this textbook, the 2004 classification of Palmini et al. was considered most apt (32). Under that system, FCD was divided into two tiers and included 4 entities: FCD Ia, Ib, IIa, and IIb. The most recent 2011 consensus on the classification of FCD has created a new third tier (FCD IIIa-d) to recognize those cases where dysplastic cortex is seen in combination with a presumed primary lesion (see below) (33).
FCDs are relatively common in surgical specimens, especially in centers with multidisciplinary epilepsy programs. The seizures associated with FCD usually manifest in the first decade. A variety of genetic predispositions and environmental insults have been hypothesized to play a role in the pathogenesis of FCD. At what time FCD arise during development is unclear (i.e., insult occurring before, during, or after neuroglial migration). Genetic studies have suggested general roles for the PI3K/mTOR and reelin pathways. In addition, polymorphisms of the TSC1 gene have been associated with FCD type IIb, while polymorphisms of the TSC2 gene have been seen with ganglioglioma (GG) and FCD type IIa. Identification of these lesions has been facilitated by MRI (34), and although a number of relatively nonspecific changes can be seen, the “transmantle sign” (involving T2/flair signal hyperintensities trailing down from the abnormal cortex and through the white matter toward the ventricular surface) is a fairly specific feature of FCD.
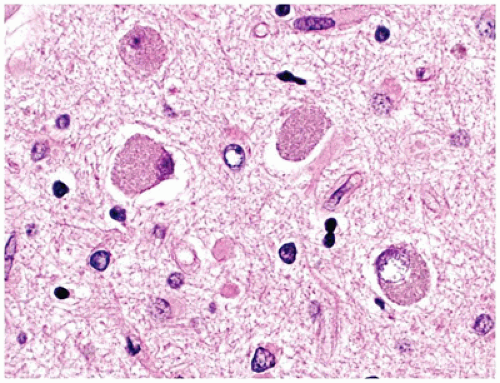
Pathologically, FCD may be grossly inapparent or seen as a focal thickening of gray matter with blurring of the underlying gray-white matter junction. According to the newest 2011 consensus paper regarding classification, FCD I is primarily categorized by abnormalities of radial migration (i.e., excessive column formation), FCD Ia, and/or tangential (i.e., dyslamination), FCD Ib, organization or a combination of both, FCD Ic. Microcolumns of FCD Ia are defined by the presence of at least 8 vertically oriented small diameter neurons that are arranged perpendicular to the cortical surface. In FCD Ib, the normal hexalaminar appearance of the neocortex is disturbed, either transcortically or focally (e.g., laminae II or IV can be missing or are significantly deplete of neurons). In both FCD Ia and Ib, (a) the gray white junction can be blurred by heterotopic neurons, (b) immature small diameter neurons can be present, and (c) hypertrophic neurons can be identified outside of layer V. The hallmark of FCD II is the dysmorphic neuron. Dysmorphic neurons are maloriented and/or abnormally large neurons with atypical coarse Nissl substance and thick dendritic processes. Cytoplasmic vacuolation can be seen. These neurons are highlighted by nonphosphorylated neurofilament protein (NFP) IHC or by Bielschowsky silver staining. Balloon cells have abundant glassy eosinophilic “astrocyte-like” cytoplasm and eccentrically placed “neuron-like” vesicular nuclei, often with prominent nucleoli; larger than gemistocytes, these cells may demonstrate neuronal, glial, or hybrid features by routine staining and by IHC (e.g., coexpressing GFAP and neuronal markers). Balloon cells, which are at times multinucleate and clustered, can be seen throughout the neocortical gray matter but most often aggregate in the subcortical white matter resulting in blurring of the gray-white junction (Figure 10-16). Dysmorphic neurons are seen in both FCD IIa and IIb, but balloon cells are restricted to FCD IIb (i.e., “Taylor’s type” FCD). The dyslamination of FCD II is different from FCD I; in FCD II, all neocortical lamina are obscured with the exception of layer I. However, both may have heterotopic neurons in layer I or the white matter, the latter resulting in blurring of the gray-white junction. Other changes may be numerous but often include gliosis, hypomyelinated zones, an abnormally myelinated layer I, and calcification (6).
FIGURE 10-16 • FCD type IIb. Balloon cells are seen within the gliotic and calcified white matter immediately subjacent to malformed cortical gray matter.
FCD type IIb is often indistinguishable from the tubers of tuberous sclerosis (TS) and as such may represent a form fruste of this condition, a hypothesis that is somewhat strengthened by genetic studies (see above). The histologic changes of FCD may also be seen in hemimegalencephaly, an epileptogenic disorder that describes the syndromic or isolated occurrence of an enlarged abnormal hemisphere associated with hemiparesis and developmental delay (6). However, the scarcely reported histology of fetal hemimegalencephaly seems more subtle and typified by accelerated cortical differentiation.
Antenatal Disruptive Lesions
Although in some sense, this group of lesions could be considered malformative; they are generally considered to result from a hypoxic-ischemic insult to the developing brain. These insults are acquired in utero and intrauterine infections may play a role in the etiology of some. Hydranencephaly is the severe and diffuse necrosis of the cerebral mantle and deep gray (with concomitant HCP ex vacuo) due to perfusion failure of the internal carotid territory at 15 to 16 weeks’ gestation. The residual mantle is markedly thinned, and there is evidence of secondary brainstem and spinal cord atrophy. There may be sparing of parenchyma supplied by the posterior cerebral artery. Porencephaly describes the focal transmantle necrosis of the cerebrum (most often in the middle cerebral artery territory) wherein the ventricle communicates with the subarachnoid space. Polymicrogyria, gliosis, and calcification often rim the porencephalic defect. If there is bilateral middle cerebral artery damage that spares the cingulate gyri (i.e., leaving a “handle”), the resulting defect is called basket brain. Schizencephaly describes a nontransmantle cleft in the cerebrum. Multicystic encephalopathy (MCE) is the result of a diffuse white/gray matter insult to the cerebrum, causing multifocal necrosis and cystic change (Figure 10-17). The insult in MCE is presumed to occur late in gestation since the general architecture of the brain is maintained, but some cases have been described in newborns.
Microcephaly and Micrencephaly
Microcephaly refers to a small head, whereas micrencephaly refers to a small brain. Some authors have used the term “microcephaly” interchangeably to refer to both. If one excludes brains with hypoplasia/atrophy due to other entities (e.g., HPE, antenatal disruptive lesions), the possibility of a rarely encountered primary micrencephaly should be considered.
FIGURE 10-17 • Microcystic encephalopathy (MCE). Coronal sections of the left cerebral hemisphere from this 2-month-old female reveal prominent pathology in keeping MCE. PCR testing of formalin-fixed paraffin-embedded tissues revealed the presence of HSV-2.
Hindbrain Malformations
As described above, by the 6th week of GA, the secondary brain vesicles that give rise to the cerebellum and pons (metencephalon), in addition to the medulla (myelencephalon), have begun their development. There are similarities in the development of the hindbrain and the spinal cord. The alar and basal plates give rise to the dorsal sensory and ventral motor spinal cord horns, respectively. With respect to the hindbrain, its dorsal aspect is essentially splayed out such that the motor basal plates lie medially, while the sensory alar plates lie laterally. The metencephalic alar plates fuse and give rise to the cerebellum. The Purkinje and deep gray neurons of the cerebellum arise from the ventricular zone of the alar plate, while the eventual internal granule neurons arise from the upper aspect of an alar plate derivative called the upper rhombic lip (35). A lateral to medial outward migration of granule neuron precursors has populated the EGL by 14 weeks and persists until 1 year of age. Neurons from the EGL migrate inward to their eventual destination in the internal granule layer. The flocculonodular, anterior, and posterior lobes are already identifiable by 12 weeks GA. Cerebellar folia can be seen by 20 weeks. The precerebellar nuclei (i.e., pontine, inferior olivary, and arcuate nuclei) arise from the lower rhombic lip.
Chiari and Dandy-Walker Malformations
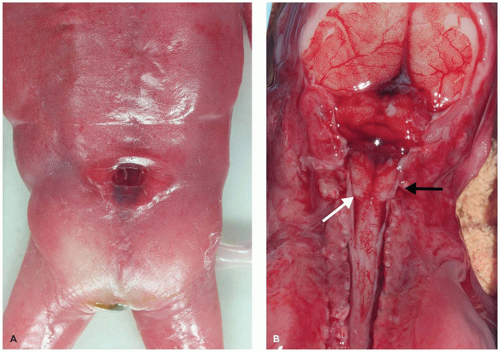
Although the Chiari malformations are discussed in the context of the hindbrain, their actual pathogenesis is unclear (see below). Three forms are well recognized. Chiari I malformations are characterized by the caudal displacement (not true herniation) of the cerebellar tonsils through the foramen magnum and into the upper cervical spinal canal. Chiari I malformations are often associated with syringomyelia. The clinical picture includes neck pain and signs/symptoms associated with syringomyelia (i.e., “cape-like” or “hanging” dissociated sensory loss in the shoulders and arms) that develops in older teenagers and young adults. Pathologically, surgical resection specimens reveal leptomeningeal sclerosis and gliosis with neuronal loss in the cerebellar cortex of the affected tonsillar tissue. Pathogenesis of the Chiari I malformation is unclear. Chiari II malformations (previously known as the Arnold-Chiari malformation) occur in young children and describe the caudal displacement of cerebellar vermis into the upper cervical spinal canal plus added hindbrain abnormalities. Ninety-five percent of Chiari II malformations are associated with a lumbosacral myelomeningocele (6). Maternal vitamin A deficiency is a risk factor for Chiari II. The clinical picture of Chiari II is dominated by HCP and the myelomeningocele. Pathologically, the fourth ventricle, midbrain, pons, and medulla are all elongated and caudally displaced. There may be “tectal beaking” and an S-shaped “kinking” of the medulla onto the dorsal spinal cord (Figure 10-18). Moreover, careful dissection of fetuses with Chiari II invariably demonstrates kinking of the medulla without cerebellar herniation upon midgestational termination, suggesting that medullary kinking precedes cerebellar herniation in the natural history of this disorder. With respect to the skull, the posterior fossa is small and there are often abnormally rostrally coursing cranial nerves. Additional CNS malformations may include PH, polymicrogyria, and pachygyria. Pathogenesis of the Chiari II malformation is unknown. Hypotheses include (a) hydrodynamic (related to mechanic pressure from HCP or excess CSF egress from the myelomeningocele), (b) cord tethering, (c) a defect in neurulation, or (d) a defect in the posterior fossa mesenchyme leading to restricted cerebellar growth. Since myelomeningoceles invariably accompany Chiari II, some have suggested that the NTD causes the brain findings characteristic of Chiari II due to egress of CSF through the NTD. To the latter, recent studies from Children’s Hospital of Philadelphia (CHOP) have demonstrated that fetal surgery to close the associated NTD helps ameliorate later clinical symptomatology (17). Chiari III malformations are rare and are defined by an occipitocervical encephalocele (which includes cerebellar tissue) that is accompanied by a distorted brain stem and abnormal local anatomy. The clinical picture is similar to Chiari II but is more severe and the prognosis is poor. Pathologic changes may include cerebellar dysplasia. Pathogenesis is also unclear but is likely related to defective neurulation.
FIGURE 10-18 • Chiari II malformation and related lumbosacral neural tube defect in a 20- to 21-week gestational aged female fetus. A: Gross examination confirms the presence of a neural tube defect, which upon further sampling for microscopy confirmed the presence of an intact spinal cord, suggesting the presence of a meningocele. B: As is typical of this gestational age, herniating cerebellar vermis is not detected in this posterior dissection; rather, a kinked medulla (white arrow) is appreciated herniating through the foramen magnum and past the C1 vertebra (the latter is dissected away and its stump is indicated by the black arrow).
The Dandy Walker Malformation (DWM) typically presents sporadically in infancy as an isolated finding or in association with other CNS malformations. Common clinical features of DWM include HCP and increased ICP. Surprisingly, cerebellar signs/symptoms are less common, but there may be concomitant cognitive impairment. DWM is characterized by five main pathologic features. First, there is cystic dilatation of the fourth ventricle. Second, the cerebellar vermis is hypoplastic or absent. Third, DWM is characterized by a large posterior fossa. Fourth, and likely related to the large posterior fossa, is the elevation of the tentorium and related dural sinuses. Finally, there is HCP. The Dandy-Walker variant has only some of the features of the DWM, which includes an anteriorly rotated vermis with or without fourth ventricular dilatation. The pathogenesis of the DWM is unknown, but maldevelopment of the fourth ventricular roof and its outlet foramina (especially Magendie) is thought to be important. Maternal isoretinoin use is a risk factor. Pathologically, the main findings are those seen grossly (see above). The fourth ventricular cyst wall is composed of an outer pial and inner ependymal layers, with residual cerebellar parenchymal in between.
A variety of additional cerebellar malformations has been characterized. Cerebellar heterotopia and dysplasia are reviewed by Golden (6). Rare malformations include cerebellar agenesis, Joubert syndrome, pontoneocerebellar hypoplasia, and granular cell aplasia. Blake pouch cyst is an emerging entity causing HCP and occasionally delayed neurologic development. All ages can be affected. Etiologically, it is thought to be due to nonperforation of the foramen of Magendie (36). Grossly, a Blake pouch cyst results in an enlarged 4th ventricle and possibly a degree of vermal hypoplasia or anterior rotation and displacement. Histologically, the wall of a Blake pouch cyst is characterized by leptomeninges, ependyma, and choroid plexus.
Malformations of the brain stem are numerous but rare. Some are described elsewhere in this chapter (i.e., Moebius syndrome, X-linked HCP with congenital absence of the pyramids). Olivary heterotopia and dysplasias of the dentate nucleus and inferior olive may occur in association with a number of different CNS malformations or syndromes, and in light of their origin from the metencephalic alar plate, it is not surprising that these may occur in conjunction with cerebellar abnormalities. Dentato-olivary dysplasia (DOD) can be seen in many clinical contexts and takes on a variety of pathologic forms. Of note, DOD can be seen in the context of “intractable epilepsy in infancy,” where pathologically it takes on a distinctive appearance characterized by a primitive C-shaped inferior olive and globular dentate nucleus (37).
TABLE 10-1 CNS CYSTS
Cyst Type
Common Sites
Pathology
Neurenteric (i.e., endodermal)
Intradural, extramedullary, and ventral to the cervical spinal cord
Cuboidal to columnar respiratory-type or GI-type epithelium covering a connective tissue stroma. Possible goblet cells and cilia. Immunohistochemistrya (IHC)
Colloid
Anterosuperior third ventricle near the Foramen of Monroe
Simple columnar epithelium. Possible cilia. Cyst contents PAS positive. IHCa
Rathke cleft
Sella
Similar to neurenteric. Degenerate forms with atrophic epithelium and xanthogranulomatous inflammation. IHCa
a Cytokeratin and EMA positive, with collagen IV immunoreactive subepithelial basement membrane. Usually CK7 positive, CK20 negative.
Cystic Lesions of the CNS
“Cysts” within the CNS are biologically benign and nonneoplastic. Their characteristic sites and pathology are summarized in Table 10-1. During embryologic development, ectopic placement of germ layer tissue may account for the formation of many of these lesions.
METABOLIC, NEURODEGENERATIVE, AND MISCELLANEOUS DISORDERS
Lysosomal Storage Disorders
Lysosomes are the digestive organelles of the cell. They contain numerous hydrolytic enzymes (e.g., phosphatases, nucleases, glycosidases, proteases, sulfatases, phospholipases) that assist in normal cellular metabolism. These enzymes are often directed to cleave off the sugar chains from larger macromolecules. A deficiency in one or more of the glycoprotein enzymes results in a lysosomal storage disorder (LSD). The LSDs are uncommon disorders that often result in progressive and multisystemic diseases that are fatal in childhood. LSD can be subdivided into categories, which are roughly based on the class of macromolecule that is not correctly metabolized; these include (a) sphingolipidoses (lipids), (b) mucopolysaccharidoses (MPS) (disaccharide molecules of glycosaminoglycans [GAGs]), (c) glycoproteinoses (glycoproteins), and (d) a number of miscellaneous LSDs, which include the neuronal ceroid lipofuscinoses (NCL) and Pompe disease (type II glycogenosis) (38).
The LSDs are generally autosomal recessive disorders that are usually the result of a mutation in the gene that encodes a particular lysosomal enzyme. There is extensive clinicopathologic overlap among the LSDs as a whole, many of which are individually indistinguishable without ancillary biochemical and genetic testing (see Chapters 5 and 28).
Conceptually, the LSDs can be divided into four basic clinicopathologic phenotypes: (a) neuronal lipidoses, (b) leukodystrophies, (c) storage histiocytoses, and (d) MPS (or the Hurler phenotype) (38). Most LSD storage products are water soluble and thus are washed out during routine histologic processing, whose residual feature is clear, vacuolated, and distended cytoplasm of involved cells. This accumulated material mechanically disrupts cellular processes and eventually leads to cell death. The neuronal lipidoses are characterized by substrate storage in cytoplasm of neurons, leading to the gross finding of megalencephaly early in the disease course (Figure 10-19). Subsequent neuronal death and gliosis eventually result in cerebral atrophy. Involvement of the retina may lead to the characteristic “cherry-red spot,” while other clinical manifestations of the neuronal lipidosis (NL) phenotype include psychomotor impairment and dementia, loss of acquired motor and perceptual skills, epilepsy, and myoclonus. The leukodystrophies, which are part of the LSDs (e.g., metachromatic leukodystrophy [MLD] and Krabbe leukodystrophy [KLD]), are the result of substrate accumulation in oligodendrocytes and Schwann cells, causing loss of myelin and myelinating cells. Clinical manifestations include psychomotor retardation, spasticity, ataxia, visual abnormalities, and a demyelinating peripheral neuropathy. Substrate accumulation in mesenchymal and epithelial cells, in addition to the extracellular matrix, results in the Hurler phenotype. Clinically, these patients have core features, which include coarse facies, skeletal and joint abnormalities (i.e., dysostosis multiplex and arthropathies), organomegaly, cloudy corneas, cardiovascular disease, and CNS disease (entrapment neuropathies, HCP, and NL). Finally, substrate storage in monocytes/macrophages causes the storage histiocytosis (SH) phenotype, which clinically manifests in hepatosplenomegaly, and hematologic and skeletal abnormalities. Table 10-2 lists a subset of the LSD, their specific enzymatic defects, and some of their characteristic clinicopathologic features (note: MLD and KLD are further described below) (see Chapter 5).
FIGURE 10-19 • Neuronal lipidosis (NL). The cytoplasm of these neurons is markedly distended by lipofuscin-like storage products in this example of neuronal ceroid lipofuscinosis (NCL).
Leukodystrophies
The leukodystrophies are a group of genetically based progressive disorders that share common abnormalities in myelin formation and metabolism. These disorders have hence been referred to as dysmyelinating, to distinguish them from acquired demyelinating disorders (e.g., multiple sclerosis) where myelin is thought to form normally but is later destroyed. Pathogenetically, the leukodystrophies are a heterogeneous group of disorders, which, for example, include some of the lysosomal and peroxisomal storage disorders. These disorders often have onset during childhood, but adults can also be affected. Clinically, they can affect numerous neurologic modalities and hence result in a myriad of signs and symptoms, which may include psychomotor impairment and dementia; pyramidal and extrapyramidal manifestations including spastic paraparesis, ataxia, and visual and hearing abnormalities; as well as signs of bulbar involvement. Characteristic clinical manifestations (i.e., age of onset, signs/symptoms) may accompany specific forms of leukodystrophy. The white matter of not only the CNS but also the PNS [e.g., MLD, KLD, and, less so, adrenoleukodystrophy (ALD)] may be affected. Some leukodystrophies are more systemic in nature and thus bear extra-CNS/PNS disease manifestations (e.g., adrenal and testicular involvement with ALD; biliary and renal involvement with MLD). Genetically, many of these disorders are inherited in an autosomal recessive manner; however, some follow an X-linked or sporadic pattern.
Pathologically, the leukodystrophies characteristically cause bilaterally symmetric white matter-predominant disease that can involve the cerebrum, brain stem, cerebellum, and even the spinal cord. Usually, subcortical U-fibers are spared from myelin destruction (Figure 10-20A). There may be a rostral (Alexander disease, MLD) or caudal (ALD; KLD) predominance of cerebral white matter disease. In general, early stages of disease are characterized by widespread myelin destruction with relative axonal preservation; macrophages are often present and distended by bubbly periodic acid-Schiff (PAS)-positive cytoplasmic material. Later stages often demonstrate axonal and oligodendrocyte destruction plus reactive gliosis. Characteristic pathologic changes often accompany individual leukodystrophies and hence assist in diagnosis (Table 10-3; Figure 10-20B). A subset of leukodystrophies are considered “sudanophilic” since macrophages and other cells that accumulate indigestible substrates stain positive with Sudan B or Oil Red O fat stains. Included in these sudanophilic leukodystrophies are ALD, Pelizaeus-Merzbacher disease (PMD), and a host of less well-described entities that may contain characteristic histopathology including calcification, pigmentation, meningeal angiomatosis, and cavitation with oligodendrocyte proliferation (e.g., vanishing white matter disease), the latter of which recently has been linked to mutations in any of the genes encoding subunits of the eukaryotic translation factor eIF2B (39,40).
Peroxisomal Disorders
Peroxisomes are cellular organelles that have a single membrane enclosing a matrix wherein biochemical reactions take place. Peroxisomes generate hydrogen peroxide (H2O2), a molecule that assists in oxidizing several cellular toxins. However, H2O2 can itself be toxic; hence, peroxisomes contain catalase, an enzyme that serves to break down H2O2 into water and oxygen. Peroxisomes also play an important role in the β-oxidation of very-long-chain fatty acids (VLCFAs), plasmalogen biosynthesis (an important cell membrane and myelin component), cholesterol biosynthesis, and the metabolism of amino acids, bile acids, and purine nucleotides. Knowledge of these basic biologic functions is clinically useful since the routine laboratory workup of the peroxisomal disorders often involves initial assessment of VLCFAs, hepatic peroxisomes, and red blood cell plasmalogens.
Palmitoyl protein thioesterase (NCL1), tripeptidyl peptidase (NCL2)
Saposin A&D (NCL1), SCMAS (subunit C of mitochondrial ATPase synthase), (NCL2-4)
NL. EM: granular osmiophilic deposit (NCL1), curvilinear bodies (NCL2), fingerprint bodies (NCL3), or “mixed” with lipofuscin-like (NCL4)
Pompe disease (glycogenosis type 2)
α-Glucosidase (acid maltase)
Glycogen (membrane bound and free by EM)
Vacuolar myopathy, cardiomegaly, macroglossia
EM: in general, many of these disorders contain membranous cytoplasmic or zebra body inclusions within lysosomes; GM1 may contain added reticulogranular material; Gaucher disease exhibits tubular inclusions; Farber granulomatosis features “banana bodies.”
FIGURE 10-20 • Leukodystrophy. A: Coronally sectioned case of Krabbe disease demonstrates symmetric dysmyelination of cerebral white matter with relative sparing of the subcortical U-fibers. (Image courtesy of Dr. Barry Rewcastle.) B: LFB-PAS-stained case of ALD demonstrates pale white matter and characteristic perivascular lymphocytic cuffing.
Deficiency of a peroxisomal ATP-binding cassette transporter, resulting in accumulation of VLCFAs. X-linked (Xq28)
Perivascular lymphocytic inflammation.
EM: trilaminar inclusions. Striated lamellar cytoplasm inclusions in CNS and select systemic organs
Metachromatic leukodystrophy (MLD)
Aryl-sulfatase A deficiency. Accumulate sulfatide (22q13)
Metachromatic material (using acidic cresyl violet or toluidine blue) in the brain (macrophages), PNS (Schwann cells), and viscera (biliary epithelium and renal tubules). EM: herringbone, prismatic, and tuft stone inclusions
Globoid cells, often perivascular. PNS: hypertrophic neuropathy with fibrosis and “onion bulbs.” EM: tubular inclusions
Alexander disease
Sporadic mutation of the GFAP gene (17q21)
RF accumulation, especially in perivascular and subpial locations. Grossly cavitated white matter. EM: amorphous electron-dense material surrounded by 10-nm intermediate filaments
More central aspects of central myelin lost with relative oligodendroglial and axonal sparing. Vacuolation at neocortical gray-white junction. No macrophages and little gliosis (versus other LSDs). EM: myelin splitting at intraperiod line plus elongate mitochondrial with “ladderlike” cristae
Pelizaeus-Merzbacher disease (PMD)
Deficiency of normal proteolipid protein (PLP). Accumulate abnormally folded PLP in the ER. X-linked (Xq22)
There are two main categories of peroxisomal disorders: (a) peroxisomal biogenesis disorders (e.g., Zellweger spectrum, and rhizomelic chondrodysplasia punctata type 1) and the (b) single peroxisome enzyme-protein deficiencies (e.g., X-ALD, D-bifunctional protein deficiency, and adult Refsum disease) (38). In general, these disorders are neuropathologically characterized by NMD, leukodystrophy-like white matter abnormalities, CNS lipid deposition, and systemic abnormalities (including the adrenal cortex and liver). Both the biogenesis disorders and the single enzymes deficiencies are inherited in an autosomal recessive fashion, with the exception of X-lined ALD. The biogenesis disorders involve mutations in the PEX genes, which encode the peroxin proteins that are important to peroxisomal functioning. Zellweger spectrum includes three disorders, which are considered to form a spectrum of diseases related to mutations in a number of genes, including PEX1. These include (from most to least severe) the following: Zellweger syndrome, neonatal ALD, and infantile Refsum disease. Zellweger syndrome (a.k.a. cerebrohepatorenal syndrome) is a systemic disorder primarily affecting the liver (cirrhosis) and brain. Patients have dysmorphic facies and neurologic manifestations that include psychomotor impairment, hypotonia, depressed deep tendon and Moro reflexes, seizures, and nystagmus. These infants die within the first year of life. Neuropathologic findings include NMDs (e.g., pachygyria, polymicrogyria), leukodystrophy-like white matter abnormalities, abnormalities of rhombic lip-derived structures (dentate nucleus/inferior olivary dysplasias, cerebellar heterotopias), and prominent deposition of sudanophilic lipid in the CNS (primarily within macrophages and showing trilaminar appearance on EM) (Chapters 5 and 15).
Mitochondrial Disorders
The mitochondria are the “powerhouse” of the cell. They produce the energy needed for life in the form of ATP via aerobic respiration. The final stages of aerobic respiration are mediated by the electron transport chain, which comprises five protein complexes that are embedded within the inner mitochondrial membrane. Each of these five complexes is composed of multiple protein subunits (86 in total), most of which are encoded by nuclear DNA. The mtDNA genome is circular and double stranded and includes 16,569 base pairs. Up to ten copies of the mtDNA genome may be seen within a cell. This genome encodes for 22 tRNAs, 2 rRNAs, and 13 subunits of the electron transport chain. Genetically induced defects in the assembly or formation of the electron transport chain, or in the maintenance of the mitochondrial DNA, result in mitochondrial disorders. Dysfunction of the electron transport chain presumably results in cell death via numerous mechanisms, including energy deprivation, free radical toxicity, and apoptosis. Since electron transport chain functioning relies on proteins encoded by both mitochondrial and nuclear DNA, mitochondrial disorders may be inherited via either maternal or classic Mendelian patterns.
Mitochondrial disorders, as with the LSDs, may display significant clinicopathologic overlap. Many of these disorders, when viewed in isolation, may be caused by more than one mutation, and in turn, any given mutation may lead to more than one phenotype. This biologic complexity makes the diagnosis of mitochondrial disorders challenging. These disorders are often described as encephalomyopathies, since muscle (cardiac and skeletal) and brain tissues are usually affected due to their heavy reliance on mitochondrial energy production. Clinically, the presence of a mitochondrial disorder may be suspected via characteristic lab abnormalities, which often include an elevation in blood/CSF lactate and the lactate-to-pyruvate ratio. Ragged red fibers (RRF) are a common manifestation of muscle disease and represent a localized proliferation of abnormal mitochondria. RRFs are detected histochemically on frozen sections via modified Gomori trichrome (dark red) or succinic dehydrogenase (dark blue) stains (Figure 10-21A). The cytochrome oxidase C (COX) stain often fails to stain such affected fibers (i.e., “pale fibers”). By EM, the abnormal mitochondria of RRFs may take several unusual configurations including concentric spirals and rectangular paracrystalline arrays that resemble “parking lots” (Figure 10-21B). It is important to note that RRFs are not entirely specific for the mitochondrial disorders and that not all mitochondrial disorders bear RRFs. Other common neuropathologic findings seen among the mitochondrial disorders include hypoxic-ischemic-like and infarct-like changes, intramyelinic edema/spongy myelinopathy, tract/system degenerations, and vascular mineralization (deep gray and adjacent white matter, dentate nucleus, and the brain stem). As our knowledge of mitochondrial disorders has evolved, so too has the number of better recognized individual entities (38). Below, only a few of the more common mitochondrial disorders are briefly discussed.
Mitochondrial encephalopathy with lactic acidosis and strokes (MELAS) is a maternally inherited disorder that most often results from an adenine to guanine point mutation at nucleotide 3243 of mtDNA. This mutation is within the gene that encodes the tRNA for leucine. Those afflicted are usually young, although both the age of onset and initial presentation may be quite variable. Sudden focal neurologic signs (e.g., hemiplegia, hemianopsia, seizures), migraine-like attacks, or more nonspecific symptoms (such as vomiting or a change in mental status) may be seen. Myopathic features include proximal limb weakness, fatigability, and deficits in eye movements. Episodes of such neurologic dysfunction tend to be recurrent. Pathologically, foci of necrotic brain damage resemble true infarcts; however, these lesions do not follow standard vascular distributions. The occipital lobes, deep gray matter (which also may demonstrate vascular mineralization), and cerebellum are often affected. RRFs are present.
Myoclonic epilepsy with ragged red fibers (MERRF) is another maternally inherited disorder. MERRF most often results from an adenine to guanine point mutation at nucleotide 8344 of mtDNA. This mutation is within the gene that encodes the tRNA for lysine. Like MELAS, those afflicted are often young. Clinical features include a proximal myopathy, myoclonic epilepsy, sensorineural hearing loss, cognitive deficits, short stature, and ataxia. Pathologic changes involve neuronal loss and gliosis among the dentatorubroolivary system, substantia nigra, dorsal column nuclei (gracile and cuneate), and Clarke’s column. Vascular mineralization may be noted in the deep gray matter, and muscle pathology includes RRFs.
Leigh disease (subacute necrotizing encephalopathy) is most frequently caused by germ line rather than mitochondrial mutations and, hence, usually inherited in an autosomal recessive pattern. Genes encoding subunits of the electron transport chain complexes I, II, IV, and V may be mutated, or alternatively, there may be a deficiency of pyruvate dehydrogenase. Disease onset often manifests prior to 2 years of age and includes weight loss, vomiting, psychomotor impairment, and weakness. Movement disorders, ataxia, eye abnormalities (optic atrophy, ophthalmoplegia, nystagmus), respiratory difficulties, hypotonia, and epilepsy are also often characteristic. Pathologically, the deep gray and brainstem are primarily affected by vasculonecrotic lesions. The brainstem tegmentum, inferior colliculi, and substantia nigra are characteristically affected. Grossly, these lesions are atrophic, soft, and symmetrically distributed. Microscopically, the findings resemble those seen in Wernicke-Korsakoff syndrome (although hemorrhagic features are absent and the mamillary bodies are normal). Typical lesions bear rarefaction of the neuropil with spongiosis and relative neuron preservation, foamy macrophages, and gliosis and an increased density of capillaries that is thought to result from vascular proliferation and/or neuropil collapse.
Only gold members can continue reading. Log In or Register to continue