Testicular Development and Disorders
Normal sexual development and differentiation are the result of a complex interplay of genetic, molecular, and endocrine factors necessary for the development of the genitourinary system including the kidneys as well as the adrenals. Embryologically, the urogenital ridges appear at around the 4th week of gestation and are initially devoid of germ cells. By the 5th week, primordial germ cells have migrated to the genital ridge and are arranged into the seminiferous cords. Up to the 6th week, both male and female gonads appear relatively similar. By the 7th week of gestation, the testes are formed with recognizable short, straight cellular tubules and are functioning with the synthesis of anti-müllerian hormone (AMH), müllerian inhibiting substance, or müllerian inhibiting factor (MIF) by the Sertoli cells, which develop from the somatic sex cord cells and subsequently the synthesis of testosterone by the Leydig cells, which develop from the intercordal gonadal mesenchyme in the 8th week (
1,
2). The intercordal mesenchyme is composed of cells that migrate from the mesonephric stroma and eventually differentiate into testicular stromal cells, and blood vessels in addition to Leydig cells. The produced hormones are essential as AMH plays a major role in the process of regression of the müllerian ducts, whereas testosterone is crucial for the differentiation of the wolffian ducts, which develop into the epididymis, vas deferens, and seminal vesicles. The rete testis develops from the mesonephric remnants in proximity to the seminiferous cords. Development of a dense fibrous tunica albuginea in the 8th week of gestation is definitive for testis formation. Testosterone synthesis peaks at 12 to 16 weeks of gestation, allowing for male secondary sexual development (
3). The remaining structures of the male genital system are derived from the urogenital sinus through the differentiation of the endoderm-derived epithelium into prostate, urethra, bulbourethral, and periurethral glands. In contrast, the wolffian duct derivatives are of mesodermal origin. The differentiation of the wolffian ducts occurs under the influence of testosterone secreted by the ipsilateral testis. The differentiation of the urogenital sinus into male external genitalia occurs under the influence of dihydrotestosterone (DHT), which is derived from testosterone by enzymatic conversion by 5α-reductase. Two additional hormones, FSH (follicle-stimulating hormone) and LH (luteinizing hormone), play important roles in the development of the male genital system mainly in the last months of gestation, regulating androgen production and Sertoli cell activity (
4,
5). The actions and the timing of these hormones are closely and precisely coordinated during development (
6). After birth, the testis continues to develop until puberty with changes affecting all testicular components until puberty.
Chromosomal sex (genotype) is established at fertilization, and from the bipotential gonads, the male genotype (46,XY) leads to the development of the testis through a series of sex chromosome-linked and autosomal genes-mediated functions. As described earlier, the testis in turn secretes essential hormones for the development of the external male genitalia (phenotype) (
7). In a normal 46,XX female, ovarian and müllerian duct development occurs because of absence of the Y chromosome, which is instrumental in the suppression of the female genitalia and reproductive organs.
Of the genes involved in the formation of the bipotential gonads,
WT1 (Wilms tumor gene),
NR5A1 (nuclear receptor subfamily 5, which encodes the steroidogenic factor 1,
SF-1), and
LIM1 are perhaps the most important (
7,
8). These events are triggered by
SRY (sex-determining region of the Y) located on the distal tip of the short arm of the Y chromosome (
8,
9). The
SRY gene encodes a protein that acts on the HMG (high mobility group) DNA-binding domain in somatic cells in the urogenital ridge to differentiate into Sertoli cells, the first differentiated cell type of the testis (
7). Although
SRY is the essential determinant of testicular development, various autosomal genes, downstream from
SYR, are involved in this process including
WT1,
SF-1,
DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1), and
SOX9 (SRY-box 9) (
10).
Congenital and Developmental Anomalies
Congenital or developmental anomalies affecting the testis may be an isolated finding or may be associated with a sexual maldevelopment syndrome that may or may not be associated with an underlying cytogenetic defect. Clinically, these conditions most commonly present as an “undescended” testis or ambiguous external genitalia.
Monorchidism, or the presence of a single testicle, may be due to a testicular regression syndrome (TRS) (discussed later), vascular injury, cryptorchidism, or another congenital anomaly. Polyorchidism (or duplication of the testis), defined as the presence of more than two testes, is a rare condition with about 100 cases reported in the literature. This condition usually manifests as triorchidism, but bilateral duplication has been also reported (
11). Clinically, two masses within the hemiscrotum, inguinal swelling and an undescended testis (UDT), or pain are the usual presentations. Associated conditions include those related to anomalies in the processus vaginalis (hernia, hydrocele), testicular maldescent, intermittent torsion, epididymitis and varicocele, as well as malignant neoplasms (
12). The exact mechanisms behind this condition are unknown, but the supernumerary or duplicate testis is suggested to result from an abnormal division of the urogenital ridge before the 8th week of gestation (
13). According to Leung’s anatomical classification, the most common variant (type II) is that of a supernumerary testis draining into the epididymis of the primary or usual testis, and hence, they share a common vas. Other variants include a supernumerary testis without any epididymis or vas and with no attachment to the usual testis (type I), supernumerary testis that has its own epididymis and both epididymides of the ipsilateral testes drain into one vas (type III), and a completely independent supernumerary testis with its own epididymis and vas (type IV) (
13). The supernumerary testis is often smaller than a normal testis, and its torsion is more frequent. The histologic appearance of the supernumerary testis ranges from normal testicular tissue with intact spermatogenesis to disorganized seminiferous tubules with diminished spermatogenesis. Rare associated chromosomal anomalies have also been reported (
12).
Cystic dysplasia of the testis (CDT) is a rare congenital malformation, presenting as testicular enlargement due to cystic dilation of the rete testis (
14). It is thought to result from an embryologic defect around the 5th week of gestation preventing the connection of the rete testis (afferent seminiferous tubule derived) with the efferent tubules (mesonephric or wolffian derived), which further drain into the epididymis. The failure of this anastomotic connection leads to cystic dilatation of rete testis in the mediastinum testis with progressive dilation of cysts compressing and replacing the adjacent testicular parenchyma. A second alternative theory of overproduction of fluid by rete testis epithelium has also been proposed (
14). Ipsilateral renal agenesis and multicystic dysplasia of the kidney are the most common associated findings with CDT. Histopathologic features of CDT include multiple, anastomosing, irregular cystic spaces of varying sizes and shapes predominantly located in the region of the mediastinum testis but also displacing the testicular parenchyma, which becomes subsequently compressed under the tunica albuginea. The cystic lining is flat epithelium resembling that of the rete testis (
15). Spontaneous regression of this lesion after conservative management has very rarely been reported in the literature (
14).
Prepubertal macroorchidism is an idiopathic condition in most cases but has also been associated with McCune-Albright syndrome, juvenile hypothyroidism, continuous splenogonadal fusion, and fragile X syndrome. Histopathologic findings may include some enlargement of the seminiferous tubules and thickening of the tubular basement membrane. The tubules contain Sertoli cells, and scattered Leydig cells are usually present in the interstitium. Focal mild interstitial fibrosis, occasional tubules containing only Sertoli cells, and focal paucity of Leydig cells are some of the pathologic findings. Some have attributed the testicular enlargement in the absence of other findings to interstitial edema and obstruction.
Vascular anomalies of the testis are rare and include angiomatous malformations and congenital lymphangiectasis. Angiomatous malformation is a rare cause of testicular enlargement and consists of numerous thin-walled vascular channels. Congenital lymphangiectasis has been reported in association with bilateral cryptorchidism and with Noonan syndrome, manifested by numerous ectatic and irregular lymphatic channels with frequent anastomoses among and around the seminiferous tubules. The seminiferous tubules may have smaller diameter, immature Sertoli cells, reduced spermatogenesis, and peritubular fibrosis (
16). Similar findings, however, have also been reported in the testes of infants without other abnormalities at autopsy.
Cryptorchidism or UDT, where either one or both testes fail to migrate to the base of the scrotum, is one of the most common genitourinary disorders in male children. The exact incidence of this condition has been reported as 1% to 5% of full term and 9% to 30% of premature males at birth (
17,
18,
19). Based on many recently published series, there has been an increase in the incidence of cryptorchidism over recent decades in North America and Europe (
20). The cryptorchid testis can be found in any position along its usual line of descent including intra-abdominally; however, approximately 80% will be located in the inguinal region, just outside the inguinal canal, and 20% to 27% of cryptorchid testes are not palpable. Rarely, ectopic locations outside the normal line of descent have been reported that included the perineum, base of the penis, abdominal wall, pubic region, and upper thigh. The majority of undescended testes will achieve full spontaneous descent by 1 year of age, predominantly within the first 3 months, and only 0.8% of infants would have incomplete descent 12 months after birth. Spontaneous testicular descent after the first year is very unlikely (
19). A number of studies have reported an
increased rate of UDT with low birth weight, maternal preeclampsia, mild gestational diabetes, and breech presentation (
21). Bilateral testicular maldescent was reported in as high as 39% of babies with cryptorchidism, which was also more likely to occur in babies with low birth weights.
In addition to the presence of a patent processus vaginalis in most cryptorchid testes other abnormalities can occur in these patients especially those pertaining to the genitourinary tract, which include dysgenetic testis, duplication of the ureter, hypospadias, renal dysplasia, and inguinal hernia (
22). Of the most common associations with cryptorchidism are malformations in the paratesticular structures such as the epididymis and its attachment to the vas deferens and gubernaculum. Epididymal abnormalities have been reported in less than 5% of normal male fetuses compared to 35% in children with cryptorchidism. The most common of these anomalies is detachment between the epididymis and the testis followed by separation of the epididymis from the vas deferens and the long looping epididymis.
Outside the genitourinary tract, abnormalities may include gastroschisis, omphalocele, imperforate anus, cardiac anomalies, lower limb anomalies, and caudal spinal malformations. It has been reported that the etiologies of cryptorchidism and hypospadias are partly shared, and the presence of both conditions simultaneously is associated with increased risk for ambiguous external genitalia and intersex disorders (
22).
Multiple causes for testicular maldescent have been suggested including an abnormal differentiation of the male sexual organs, midline abnormalities, anatomical anomalies of the gubernaculum testis, hormonal dysfunction affecting the hypothalamic-pituitary-testicular axis (hypogonadotropic hypogonadism), mechanical impairment (insufficient intraabdominal pressure, short spermatic cord, underdeveloped processus vaginalis), and heredity.
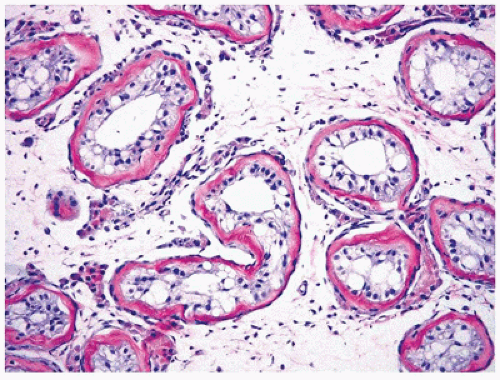
The prepubertal UDT is usually smaller than the contralateral one, and the difference becomes even more significant with progressive lesions. Generally, there is correlation between the age at the time of orchiopexy or orchiectomy and the severity of the histologic alterations seen in the testis. The isolated cryptorchid testis in the prepubertal child has only subtle quantitative abnormalities such as decreased percentage of tubules containing germ cells (low tubular fertility index), decreased mean tubular diameter, apparent tubular loss, and early interstitial fibrosis (
23) (
Figure 19-1). The postpubertal and adult cryptorchid testis usually exhibits abnormalities in all testicular structures characterized by tubular sclerosis, maturational arrest in spermatogenesis, and interstitial fibrosis. Tubules with immature Sertoli cells only (Sertoli cell nodules) may also be found (
Figure 19-2), and areas of Leydig cell hyperplasia are frequent. Granular transformation and degeneration of Sertoli cells have been reported in the cryptorchid testis as well as in other testicular disorders (
24). Similar histopathologic features may also be found in retractile testis (which is essentially a suprascrotal testis but can be easily manipulated into the scrotum), indicating that these conditions may share some causal relationships and that they may require similar management approach (
25). Some changes in the rete testis have been reported particularly in postpubertal cryptorchid testis including adenomatous features or dysgenesis with hypoplastic changes.
The relationship between cryptorchidism and male infertility has been extensively studied, and links between the two conditions have been made in a number of series. Cryptorchidism has been reported as the cause of infertility in up to 9% of cases. Biopsies from cryptorchid testes may reveal lack of germ cells as early as 18 months of age, the incidence of which increases with advanced age and with bilaterality (
26). Similar trends were observed even in patients who underwent orchiopexy, implying that the actual development of germ cells in cryptorchidism might also be impaired (
26). Some authors, however, cast some doubt on the level of certainty of this causative relationship between cryptorchidism and infertility and advocate that while it is certain that untreated men with bilateral abdominal testes will be infertile, the levels of fertility are unpredictable in other less severe scenarios (unilateral cryptorchidism, inguinal testes, postorchiopexy).
Another important association with cryptorchidism is the increased risk of developing testicular germ cell tumors (GCT), especially seminoma, compared with normally descended testes (
18). The relative risk of GCT in a cryptorchidism patient is approximately 2.75 to 8. The relative risk of GCT is between 2 and 3 in patients who undergo orchiopexy by ages 10 to 12 years. However, patients who undergo orchiopexy after age 12 years or no orchiopexy might be two to six times as likely to have GCT as those who undergo orchiopexy at a prepubertal stage. Multiple studies in literature demonstrate conflicting opinions, but overall, it seems that a contralateral, normally descended testis in a patient with cryptorchidism carries no increased risk of GCT. It has
been demonstrated that the risk of developing a malignancy increases with an abdominal testis location compared with an inguinal testis and also with those treated with orchiopexy postpubertally (
26). In the latter case, the tubules are arrested in maturation. Interestingly, persistently cryptorchid testes at inguinal and abdominal locations might be at higher risk for development of seminoma, while corrected cryptorchid or scrotal testes might be at higher risk of developing GCT of the nonseminomatous type (
18).
Disorders of Genital Differentiation
These disorders are generally associated with a normal chromosomal composition and normal gonads. This includes female and male pseudohermaphroditism.
Female pseudohermaphroditism occurs as a result of relative androgen excess
in utero in an individual with two ovaries and a 46,XX genotype. The elevated levels of androgen present during embryogenesis usually result in genital ambiguity and may result in a male phenotype. The most common cause is the
adrenogenital syndrome (AGS, congenital adrenal hyperplasia). The manifestations of AGS in genotypically female patients are related to defects in the biosynthetic pathways of mineralocorticoid, glucocorticoid, and sex steroids. In males with AGS, usually there is no evidence of genital ambiguity, but they may have an enlarged phallus. They may also develop clinically detectable bilateral testicular nodules during childhood or young adulthood that may be confused with true Leydig cell tumors (LCTs) and are designated as testicular
tumors of the AGS (
29) (see
Chapters 18 and
21).
A rare condition, placental aromatase deficiency, causes maternal virilization during pregnancy and pseudohermaphroditism of the female fetus. Due to mutations in the aromatase gene CYP19 and the resulting lack of aromatase activity, fetal androstenedione cannot be converted to estrogen by the placenta and instead is converted to testosterone peripherally, resulting in virilization of both fetus and mother.
Other conditions that might be associated with female pseudohermaphroditism are related to maternal factors such as
maternal ingestion of synthetic progestins or androgens or the presence of
maternal virilizing lesions during pregnancy including a luteoma of pregnancy (
27).
Male pseudohermaphroditism represents a heterogeneous group of intersex conditions that occur in individuals with normal 46,XY karyotype and either identifiable testes or evidence that testes were present during fetal development but the external genitalia are usually female or ambiguous (
30). The responsible defect may be (a) at the gonad level, leading to disorders of testosterone biosynthesis and metabolism or testosterone receptor abnormalities, or deficiency in MIS gene or (b) at the end organ level, where the developing tissues are unresponsive to androgen stimulation leading to an abnormal phenotype. Other less well-defined causes may also be responsible.
Gonadal defects responsible for male pseudohermaphroditism include TRS, agenesis or deficiency of the Leydig cells, defects in specific enzymes in the pathway of testosterone or DHT biosynthesis or receptors to these hormones, or a defect in elaboration or action of MIS.
Testicular regression syndrome (TRS, congenital anorchia, vanishing testis) is a condition in which a testis is
thought to have once existed but has atrophied and disappeared during early development (
31,
32). The testis is clinically impalpable, and no normal testicular tissue can be identified following exploration. Generally, congenital absence of the testis, or testicular agenesis, is an uncommon anomaly as it was detected in less than 1% of testes both in fetuses and cryptorchid patients. This condition results from the irreversible destruction of one or both testes during fetal life in an XY individual, resulting in variable hormonal deficiencies and developmental anomalies based on the stage at which testicular damage occurred (
33). Unilateral testicular destruction does not result in TRS. By histopathologic examination, the testis may be completely absent or represented by only a microscopic remnant. In addition to having no gonadal tissue, pathologic findings include a collection of vascularized fibroconnective tissue (85%), hemorrhage or hemosiderin deposition (70%), calcification (60%), or giant cells near the residual vas deferens or epididymis, the expected site of the gonad (
31,
32,
33). The vas deferens ends blindly, and a small circumscribed nodule of tissue may be located in the retroperitoneum, in the iliac fossa, or in the scrotum. By definition, no evidence of preserved remnants of seminiferous tubules should be present.
The clinical presentation of individuals with TRS is variable and is reflective of the specific stage of fetal development during which the testes were damaged. Generally, at one end of the spectrum, when gonadal regression occurs early in embryonic life before the testes release androgenic or antimüllerian hormones, the testes are absent and the phenotype is female. At the other end, regression occurring later and through fetal life would allow for a male phenotype with infantile to nearly normal male genitalia and differentiated wolffian-derived structures. Affected individuals commonly have ambiguous genitalia. A number of etiologies have been proposed for TRS including inherited genetic defect, intrauterine infection, and infarction-torsion (
27).
It is presumed that testicular regression develops late in the fetal period after the müllerian structures regressed under the influence of the müllerian inhibitory substance and the male gonads and genitalia developed under the influence of the androgens. Despite the familial occurrences of TRS suggesting a genetic etiology, no specific genes have been identified to be associated with it and in particular those related to the opening reading frame sequence of SRY. Unlike cryptorchidism, there is no increased risk of gonadal neoplasia, because there is little, if any, residual gonadal tissue.
Leydig cell deficiency (agenesis or hypoplasia) is a rare condition of male pseudohermaphroditism thought to be due to a defect in the human chorionic gonadotropin-LH receptor, primary agenesis or hypoplasia of the Leydig cells, or an abnormal LH receptor molecule. Affected individuals are genotypically males (46,XY) with female phenotype and unremarkable or ambiguous external genitalia. Bilateral, slightly small to normal-sized cryptorchid testes are present with fully or partially developed epididymides and vasa deferentia, indicating that testosterone production by Leydig cells was intact early in embryonic development. The testes exhibit interstitial fibrosis, but no mature Leydig cells are present and no testosterone production is noted. LH levels are elevated in affected individuals. Tubules with Sertoli cells are found, and müllerian structures are typically absent, indicating appropriate testicular production of MIS by Sertoli cells during fetal life (
27).
Familial occurrence of this condition has been reported, and a number of mutations in the transmembrane domain of LH receptor gene, resulting in Leydig cell deficiency, have been identified (
34).
Defects in testosterone synthesis may be due to inborn errors of the enzymes involved in testosterone biosynthesis in the testis or the adrenal gland that may result in subnormal levels of testosterone and DHT during embryogenesis (relative estrogen excess) resulting in female or ambiguous external genitalia. These defects may involve cholesterol synthesis (mutations in 7-dehydrocholesterol reductase gene) as in Smith-Lemli-Opitz syndrome or mutations in the steroidogenic enzymes responsible for the conversion of cholesterol to testosterone and DHT, which include (a) steroidogenic acute regulatory protein (StAR) gene responsible for congenital lipoid adrenal hyperplasia, (b) 17α-hydroxylase and 3β-hydroxylase dehydrogenase responsible for congenital adrenal hyperplasia, and (c) 17-ketosteroid reductase.
The degree to which the external genitalia develop depends upon the type and the severity of the defect. The microscopic features of testes in patients with these conditions vary and may show large clusters of Leydig cells surrounding seminiferous tubules. Germ cells (spermatogonia) are often normal in children but disappear by puberty resulting in Sertoli-only syndrome. Some germ cells, however, can persist and rarely develop into intratubular germ cell neoplasia. Müllerianderived structures are absent, but wolffian duct structures may be present (
27).
Defect in müllerian inhibiting system or the
persistent müllerian duct syndrome (PMDS), also referred to as
hernia uteri inguinale, is a rare form of male pseudohermaphroditism characterized by the presence of müllerian duct structures in 46,XY phenotypic males. The age at diagnosis ranges from a neonate to the fourth decade. Most patients have unilateral or bilateral cryptorchid testes, normal or almost normal male external genitalia, and an inguinal hernia containing a prolapsed infantile uterus and fallopian tubes (
35). The testes may be histologically normal, and the wolffian duct structures are developed with the vas deferens embedded in the wall of the upper vaginal structure in most cases. Inguinal hernias occur in almost 40% of cases. Malignant testicular tumors such as intratubular germ cell neoplasia and seminoma have been rarely reported in cases of adult PMDS patients with uncorrected cryptorchid testis. More recently, rare examples of clear-cell adenocarcinoma of the müllerian duct and uterine adenosarcoma in a boy with PMDS have been reported. PMDS has been reported with a familial occurrence and rarely in identical male twins.
PMDS is currently considered a heterogeneous group of disorders caused by at least two different defects in the müllerian inhibiting system. The most common is a defect in the MIS (müllerian inhibiting substance) gene, also known as AMH (antimüllerian hormone) gene preventing it from producing any biologically functional MIS. The second defect involves an abnormal AMH type II receptor resulting in end-organ insensitivity to MIS despite the presence of biologically active MIS. In other patients, an abnormality in the timing of MIS secretion may exist.
End-Organ Defects
As mentioned earlier, responsiveness to androgen is required for the normal development of the external genitalia and wolffian duct-derived structures. The presence of the enzyme 5α-reductase in the anlage of the prostate and external genitalia is also required for the conversion of testosterone to DHT. An absent or unstable androgen receptor in 46,XY individuals leads to impaired development of both wolffian duct-derived structures and external genitalia. If only 5α-reductase is absent or defective, abnormalities confined to the external genitalia and prostate will be observed.
Androgen receptor disorders (androgen insensitivity syndromes) result in variable phenotypes ranging from a female phenotype with intra-abdominal testes to ambiguous genitalia to a male phenotype with minimal clinical abnormalities.
Complete testicular feminization due to complete androgen insensitivity (e.g., testicular feminization, Goldberg-Maxwell-Morris syndrome, hairless women, androgen receptor insufficiency) is the most common form of male pseudohermaphroditism occurring in 1 of 20,000 newborns. It is caused by failure of androgen receptor binding despite its production and secretion by the fetal testis. The underlying mechanisms have been identified as mutations in the androgen receptor gene including point mutations resulting in amino acid substitutions or premature stop codons, frameshift mutations by nucleotide insertions or deletions, complete or partial gene deletion, or intronic mutations affecting the splicing of the androgen receptor RNA.
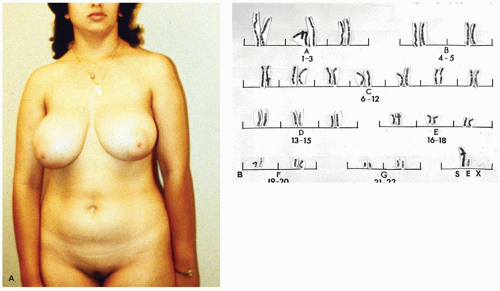
Due to the presence of phenotypically female external genitalia (
Figure 19-3), the condition is rarely diagnosed before puberty unless an inguinal hernia or labial mass is encountered or unless the disorder is known to be familial. Primary amenorrhea is the most common complaint leading to evaluation and subsequent diagnosis. The wolffian tract involutes resulting in cystic epididymides that are usually not connected to the testes. The vasa differentia, seminal vesicles, and prostate are absent. As a rule, both the cervix and the uterine corpus are absent. A fragment of fallopian tube may be found in up to one-third of cases (
27).
The testes are cryptorchid and may be intra-abdominal or inguinal, or in the labia majora, and 50% are found in inguinal hernias. Overall, the testes in androgen insensitivity
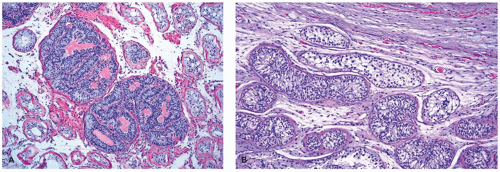
syndrome are histologically similar to the cryptorchid testis except that the tubules are less mature with possible spermatogonia but no spermatogenesis. Leydig cells are absent or replaced by collagenized interstitial tissue in portions of the gonad, whereas sheets of Leydig cells may be found near the hilus and nerves. Ovarian-like stroma replaces the testicular interstitium. Hamartomas and Sertoli cell adenomas were reported in the majority of cases in the postpubertal testis. These hamartomatous nodules are multiple, bilateral, tan, yellow, or white in appearance with bulging cut surface and may be composed of immature Sertoli cells, germ cells, Leydig cells, ovarian-type stroma, nonspecific fibrous stroma, and smooth muscle. The typical size varies from 1 to 10 mm, but may occasionally be up to 40 mm. Sertoli cell adenomas consist of nodules of predominantly or exclusively packed seminiferous tubules with immature Sertoli cells that are 3 cm in average size but range up to 25 cm. A rare example of a testicular tumor resembling the sex cord with annular tubules has been reported. GCT, particularly seminoma and less commonly intratubular germ cell neoplasia, can sometimes be encountered in patients with this syndrome, and, rarely, sex cord-stromal tumors have been reported (
27). The development of malignant gonadal tumors in patients with testicular feminization usually occurs later in adulthood.
Partial androgen insensitivity syndrome due to partial androgen receptor insufficiency accounts for 10% of all cases of androgen insensitivity and encompasses several different phenotypes, ranging from individuals with a predominantly female appearance to persons with ambiguous genitalia or individuals with a predominantly male phenotype. Affected patients typically present at birth with genital ambiguity, but severe hypospadias, micropenis, bifid scrotum, and bilateral cryptorchidism are also common. Alternatively, the external genital phenotype may be predominantly female with partial labial fusion and clitoromegaly. The underlying mechanism involves a qualitative defect in the androgen receptor. Additionally, a number of syndromes and conditions are characterized by partial androgen insensitivity including Reifenstein, Lubs, Gilbert-Dreyfus, Rosewater, and the infertile male syndromes and Kennedy disease.
A disorder of peripheral testosterone metabolism is caused by mutation in the enzyme 5α-reductase type 2, which is responsible for converting testosterone to DHT to exert its effect on differentiating the urogenital sinus into external male genitalia and prostate. Affected males usually have female to ambiguous external genitalia at birth. The penis is small (clitoris-like) and lacks a urethral orifice. A blind vaginal pouch and inguinal or labial testes may be observed. Wolffian-derived structures are normal but no müllerian-derived structures are present. Due to activation of type 1 isoenzyme, some virilization occurs at puberty demonstrated by penile enlargement, scrotal rugation and hyperpigmentation, and testicular enlargement and descent. Microscopic findings of testicular tissue may include spermatogenesis, tubular atrophy, no spermatogenesis, or Leydig cell hyperplasia. The prostate remains rudimentary, and the seminal vesicles remain underdeveloped (
27).
Disorders of Sexual Determination
These disorders are generally associated with sex chromosome abnormalities resulting in abnormal gonad formation. Affected individuals characteristically have additions, deletions, or mosaicism of the sex chromosomes, and the appearance of the gonads is variable, ranging from a streak gonad to a nearly normal female or male both grossly and microscopically.
Mixed gonadal dysgenesis (MGD) is one of the most frequent causes of male sexual ambiguity in individuals usually with a 45,X/46,XY or 46,XY karyotype. In one series, MGD was the diagnosis in approximately 8% of children with intersex conditions. It represents a heterogeneous group of abnormalities characterized by persistent müllerian duct structures, a dysgenetic testis, and a contralateral streak gonad (
30). The phenotypical heterogeneity of MGD is attributed to the presence of a variety of different genetic abnormalities causing the syndrome mostly related to deletions of both the short and the long arms of chromosome Y.
The loss of testicular functions leads to incomplete inhibition of müllerian development, incomplete differentiation of wolffian duct structures, and incomplete male development of the external genitalia. Testicular maldescent may also occur, and some patients have phenotypical features of a Turner-like syndrome. An infantile or rudimentary uterus and at least one fallopian tube are found on the side with the uncommitted streak gonad. An intra-abdominal or inguinal cryptorchid testis or fibrous streak dysgenetic testis without an accompanying fallopian tube is present on the contralateral side. Organs of wolffian duct derivation may be present with variable frequency. An epididymis is identified in twothirds of cases and is usually present on the side where there is a testis. The vas deferens is encountered less frequently, and the seminal vesicle is identified only rarely.
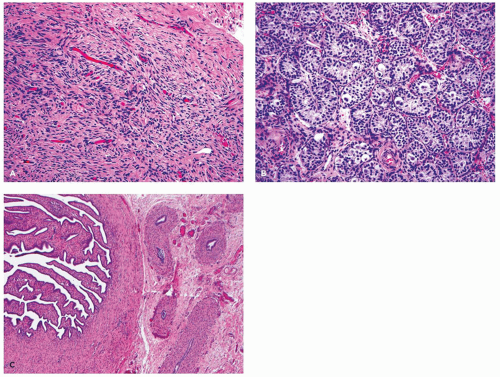
The gonad may be a testis or a streak gonad (
Figure 19-4). Streak gonads may show partial differentiation into testicular phenotype, or may exhibit features toward ovarian differentiation with the characteristic ovarian-type stroma and rare primordial follicles. However, no true ovary is present, which requires the presence of differentiation with follicles in at least the antral stage (
27). A unilateral macroscopic testis is found in 60% of cases, whereas bilateral testes may be seen in about 15% of cases usually with an asynchronous degree of maturity.
The testicular architecture is consistently abnormal in these individuals as the region of the tunica albuginea or cortex contains widely spaced seminiferous tubules with ovarian-like stroma or immature primary sex cords indeterminate between female and male structures. The medullary region may contain normal seminiferous tubules and interstitium, but in some cases, it is difficult to distinguish between female and male structures. Occasional narrow closed seminiferous tubules are lined by Sertoli cells, and in other examples, the germ cells may be seen directly lining the basement membrane of the seminiferous tubule without the Sertoli cell layer that usually normally surrounds them. Leydig cells may be present in small clusters of varying size.
Occasionally, broad zones of the cortex may exhibit a degree of differentiation toward streak-like ovary, even displaying rare primordial follicles. By puberty, the germ cells present in a streak gonad may degenerate and disappear, resulting in a gonad composed exclusively of fibrous tissue and a few rete tubules. The hilar region of the streak gonad or streak testis is populated by hilus cells and rete or mesonephric tubules. Hyperplasia of the hilus cells in response to pituitary gonadotropins may result in clinical virilization. Tumors develop in about 10% of those with MGD and the dysgenetic gonads of which 25% to 30% are malignant, the most common of which is gonadoblastoma accounting for 75% to 80% of all germ cell neoplasms in this disorder. However, other GCTs have been also reported including germinoma, yolk sac tumor (YST), teratoma, embryonal carcinoma (EC), and choriocarcinoma. These tumors may be unilateral or bilateral. Occasionally, Sertoli cell tumor (SCT) and SCT-like proliferations of sex cord elements have been also reported. Early gonadal resection is recommended in order to avoid the development of an invasive GCT and to avoid the consequences of onset of virilization in a patient who has been raised as a female.
Pure gonadal dysgenesis (PGD) refers to phenotypically female individuals with streak gonads and internal genitalia that include müllerian structures (uterus and fallopian tubes). It occurs with both 46,XX and 46,XY karyotypes and has both familial and sporadic patterns of inheritance. The stroma of the gonads has an ovarian-like appearance, and primary amenorrhea is the usual clinical presentation. PGD patients with 46,XX karyotype only rarely develop gonadal tumors, examples of which include GCT and mucinous epithelial tumors. Some have hilus cell hyperplasia and hilus cell tumors with the usual associated virilizing effects.
PGD patients with 46,XY karyotype are at higher risk for gonadoblastoma and other GCTs that may develop in 10% to 25% of cases and can be unilateral or bilateral (
36,
37).
True hermaphroditism (TH) is a disorder of gonadal differentiation defined by the concurrence of both ovarian and testicular tissue, with coexistent ovarian follicles (not just connective tissue stroma) and seminiferous tubules (not just Leydig cells). The gonads may be ovary and testis separately or combined in an ovotestis (
27,
38). Affected individuals may have either a female or a male phenotype with variable degrees of sexual ambiguity. The clinical manifestations are variable and depend on the gonadal tissue present and the age at the time of diagnosis. TH is a rare condition both in North American and Europe but is more commonly encountered in Africa, especially in South Africa.
The architecture and the distribution of gonadal tissues in TH take several forms with asymmetry of the gonads in the majority of cases. An ovotestis represents the most frequently encountered type of gonad in this condition (
38,
39,
40). Patterns of gonadal development include an ovary on one side and a testis on the other (30% of cases) or an ovary on one side and a contralateral ovotestis (30%). Bilateral ovotestes are found in 20% or more of true hermaphrodites, and a testis-ovotestis combination is found in 10% of cases. In the majority of cases (approximately 80%), the ovarian and the testicular tissues are arranged in an end-to-end fashion with a distinct line demarcating the two tissues. The ovary, which is the second most common gonad in TH, preferentially develops on the left side, whereas the testis, which is the least common gonad encountered in TH, develops preferentially on the right (
27). The location of the gonad is influenced by the type and the quantity of gonadal tissue present. Increasing amounts of ovarian tissue heightens the probability that the gonad will be in an ovarian position, and as a result, it is very unlikely for female gonadal tissue (either ovary or ovotestis) to be situated in the inguinal canal or in the labioscrotal fold. The position of the testis is less constant as most reside in the scrotum but can be encountered in the inguinal region or in the normal ovarian position. The nature of the genital structure adjacent to a gonad in TH follows that of the ipsilateral gonad, which is characterized by having a fallopian tube adjacent to an ovary and an epididymis or vas deferens adjacent to a testis. Either a müllerian (more commonly) or wolffian structure, but not both, is adjacent to an ovotestis.
In young patients, the microscopic appearance of the gonadal tissue is often normal with the ovarian tissue containing numerous follicles, whereas the testicular parenchyma has normal-appearing seminiferous tubules with spermatogonia. In older patients, ovarian tissue with structures indicative of ovulation (follicles, corpora lutea, and corpora albicantia) may be seen, but the testicular tissue (in testis or ovotestis) is usually abnormal with incomplete development, lack of spermatogenesis, loss of germ cells, and tubular sclerosis. Scrotal testes in these patients show less severe changes, sometimes showing faulty spermatogenesis (
27).
The prevalence of gonadal neoplasms, mainly gonadoblastoma and other types of malignant germ cell neoplasms, is estimated at 2% to 3% of cases (
41). A rare case of juvenile granulosa cell tumor (JGCT) in this setting has been reported.
The causes of TH are probably as varied as the karyotypic expressions, and genetic aberrations appear to play a key role in its development. Patients with a “Y” chromosome have a two – to threefold increased frequency of having a testis as opposed to an ovotestis, and nearly 75% of true hermaphrodites with an ovary and an ovotestis have a 46,XX karyotype. A 46,XX/46,XY karyotype represents true genetic chimerism, whereas the 46,XX karyotype is very likely to represent a crossing over of the X and Y chromosome during first meiotic division in the primary spermatocyte, or the presence of hidden mosaicism for
SRY (
42). There are examples where the patients were 46,XX and lacked the
SRY gene in usual cells examined (leukocytes), but cells from the gonad itself demonstrated
SRY. Autosomal dominant mutations that mimic
SRY have been suggested as one possibility where
SRY was absent. The 46,XY karyotype probably contains a hidden 46,XX cell line or that
SRY, if present, may act at a time too late to stimulate the development of a testis, hence permitting ovarian tissue to develop.
Klinefelter syndrome (KS) is one of the most common causes of prepubertal delay and primary hypogonadism in males, occurring in about 1 of every 500 to 1 of every 1000 live newborn males and accounting for about 3% of infertile males (
27,
43,
44). In the majority of cases, the karyotype is 47,XXY, which usually results from nondisjunction occurring during meiosis of either paternal or maternal gametes. The clinical picture varies depending on the age when the diagnosis is first suspected. Men with KS present with sequels of androgen deficiency like infertility, low testosterone, erectile dysfunction, and low bone mineral density. They typically are tall men with narrow shoulders, broad hips, sparse body hair, gynecomastia, small testicles, and azoospermia. Infants with KS may have normal external male genitalia at birth, which may cause a delay in its discovery. However, in some individuals, other findings may be indicative of this syndrome such as hypospadia, micropenis, and small, soft testes or cryptorchidism. In adults with KS, the testes are small and rarely exceed 2 cm in greatest dimension. Histologically, the seminiferous tubules may show some degenerative changes during fetal life, which increases with age to the point that by late childhood, the primary spermatogonia are greatly decreased in number. This degenerative process may dramatically accelerate shortly before the expected time of puberty. In adults, the testes are largely atrophic with hyalinized seminiferous tubules and prominence of Leydig cells. Some tubules may be preserved, but lined only by Sertoli cells. Functionally, the Leydig cells are abnormal, as evidenced by low levels of serum testosterone with elevated levels of serum LH and FSH.
A variety of neoplasms have been associated with KS including both gonadal and extragonadal GCTs. Most extragonadal tumors occur in the mediastinum as teratoma and EC, but rare examples of primary intrapelvic seminoma have been reported. In the testis, seminoma, teratoma, and EC have been encountered. LCTs are rare. Men with KS are at a higher risk of developing breast carcinoma than men without KS (
45). Additionally, various hematologic malignancies have been reported in individuals with KS, including acute leukemia, chronic myeloid leukemia, and malignant lymphoma.
Turner syndrome is a disorder of sexual differentiation that is discussed in detail elsewhere in the book, whereas
Turner-like mosaicism (45,X/46,XY) is part of the MGD discussed earlier (see
Chapter 18).