Chapter 15 The Kidneys and the Urinary System




















4 What are the common clinical features of uremia?
Clinical features of uremia may be found in essentially all major organs and include:






























8 What are the causes of acute renal failure?
Three types of renal failure are recognized: prerenal, renal, and postrenal. See Table 15-1.
TABLE 15-1 Cause of Acute Renal Failure
| Type of Renal Failure | Pathogenesis | Clinical Condition |
|---|---|---|
| Prerenal | Decreased renal perfusion | Congestive heart failure |
| Loss of blood | Massive bleeding | |
| Renal | Glomerular disease | Acute glomerulonephritis |
| Tubulointerstitial nephritis | Drug reaction | |
| Vasculitis | Wegener granulomatosis | |
| Toxic tubular necrosis | Mercury poisoning | |
| Postrenal | Intratubular obstruction | Acute urate nephropathy |
| Renal–pelvic obstruction | Nephrolithiasis | |
| Ureteric obstruction | Urinary stones | |
| Bladder/urethral obstruction | Prostatic hyperplasia |
9 How does chronic renal failure develop?
 Diminished renal reserve: These patients do not have clinical or laboratory signs of renal disease (BUN and Cr are normal). Additional testing will reveal that GFR is significantly reduced, and azotemia may develop during intercurrent diseases.
Diminished renal reserve: These patients do not have clinical or laboratory signs of renal disease (BUN and Cr are normal). Additional testing will reveal that GFR is significantly reduced, and azotemia may develop during intercurrent diseases.


10 Define derangements of urine volume
 Anuria: It is characterized by reduced urine output (<100mL urine per day), reflecting renal injury.
Anuria: It is characterized by reduced urine output (<100mL urine per day), reflecting renal injury.

DEVELOPMENTAL DISORDERS
15 What are the differences between autosomal dominant and autosomal recessive kidney disease?
Comparisons of autosomal dominant and autosomal recessive polycystic kidney disease are illustrated in Fig. 15-1 and listed in Table 15-2.
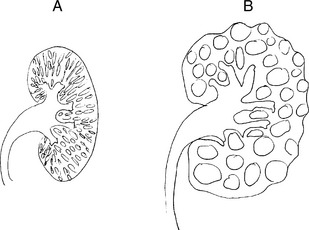
TABLE 15-2 Autosomal Dominant and Autosomal Recessive Polycystic Kidney Disease (PCKD)
| Feature | Autosomal Dominant PCKD | Autosomal Recessive PCKD |
|---|---|---|
| Incidence | Common (1:800) | Rare (1:15,000) |
| Inheritance | Autosomal dominant | Autosomal recessive |
| Gene | Polycystin genes (PKD1 = 85%) | Fibrocystin, PKHD1 |
| Bilateral | Yes | Yes |
| Gross appearance | Large cystic kidneys (>1000g) | Spongelike symmetrically enlarged (100–200g) |
| Cysts | Large (from any tubule) | Small (collecting duct derived) |
| Symptoms | Infancy, childhood | Adulthood (>35 years) |
| Associated anomalies | Polycystic liver disease (20%) Berry aneurysms of cerebral arteries | Small bile duct cysts, liver fibrosis |
GLOMERULAR DISEASES
16 What is the difference between primary and secondary glomerular diseases?
Primary glomerular diseases are, for example:



Secondary glomerular diseases occur in the course of systemic diseases such as:












Key Points: Glomerular Diseases




20 Describe two forms of antibody-associated forms of glomerular injury
 Antibodies to endogenous antigens of the GBM: This mechanism accounts for the renal injury in Goodpasture syndrome, a disease caused by antibodies to collagen type IV.
Antibodies to endogenous antigens of the GBM: This mechanism accounts for the renal injury in Goodpasture syndrome, a disease caused by antibodies to collagen type IV. Antibodies to nonglomerular antigens: Antigen–antibody complexes found in the glomeruli may result from two pathogenetic mechanisms: in situ formation or intraglomerular deposition of circulating immune complexes.
Antibodies to nonglomerular antigens: Antigen–antibody complexes found in the glomeruli may result from two pathogenetic mechanisms: in situ formation or intraglomerular deposition of circulating immune complexes. In situ immune complex formation results from the binding of circulating antibodies and the “implanted” antigen fixed to the basement membrane of the glomerulus. This mechanism accounts for the formation of glomerular lesions in poststreptococcal glomerulonephritis. It is assumed that the streptococcal antigens are implanted into the GBM during the infection, and when the antibodies are formed, they reach the implanted antigens in the glomeruli, thus forming immune complexes.
In situ immune complex formation results from the binding of circulating antibodies and the “implanted” antigen fixed to the basement membrane of the glomerulus. This mechanism accounts for the formation of glomerular lesions in poststreptococcal glomerulonephritis. It is assumed that the streptococcal antigens are implanted into the GBM during the infection, and when the antibodies are formed, they reach the implanted antigens in the glomeruli, thus forming immune complexes.
21 What pattern of staining of glomeruli is seen by immunofluorescence microscopy in anti-GBM nephritis?
22 What pattern of staining of glomeruli is seen by immunofluorescence microscopy in circulating immune complex nephritis?
24 In which parts of the glomeruli may the antigen–antibody complexes be seen by electron microscopy in various forms of glomerulonephritis?
Depending on their location, immune complexes are classified as follows:





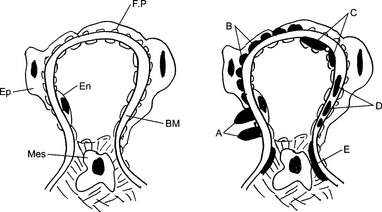
25 Why does the immunologic injury of the GBM cause proteinuria?
 Increased permeability of the GBM without interruption of the filtration barrier: It results from complex physicochemical changes caused by the action of activated complement, enzymes, and cytokines. These changes involve the biochemical composition and electric charges of the GBM and the size of internal pores that serve as normal channels for the passage of proteins.
Increased permeability of the GBM without interruption of the filtration barrier: It results from complex physicochemical changes caused by the action of activated complement, enzymes, and cytokines. These changes involve the biochemical composition and electric charges of the GBM and the size of internal pores that serve as normal channels for the passage of proteins.
27 List the soluble mediators of inflammation that contribute to the antibody-mediated glomerular injury
 Activated complement components act as chemotactic fragments and mediate the influx of neutrophils and macrophages. Complement may also increase the permeability of the GBM and cause mechanical lesions through the action of membrane attack (C5–C9 terminal complex).
Activated complement components act as chemotactic fragments and mediate the influx of neutrophils and macrophages. Complement may also increase the permeability of the GBM and cause mechanical lesions through the action of membrane attack (C5–C9 terminal complex).



28 What are the histologic signs of chronic progression of glomerular disease?
 Focal segmental glomerulosclerosis: This is a secondary change that occurs in the relatively unaffected glomeruli of a diseased kidney that has lost many nephrons. Normal glomeruli receive increased amounts of blood and undergo hypertrophy. The hemodynamic changes in the overburdened and enlarged glomeruli lead to segmental hyalinization of capillary loops and progressive narrowing of their lumen by sclerosis.
Focal segmental glomerulosclerosis: This is a secondary change that occurs in the relatively unaffected glomeruli of a diseased kidney that has lost many nephrons. Normal glomeruli receive increased amounts of blood and undergo hypertrophy. The hemodynamic changes in the overburdened and enlarged glomeruli lead to segmental hyalinization of capillary loops and progressive narrowing of their lumen by sclerosis.
29 Discuss the histologic features of acute glomerulonephritis
 Hypercellularity of glomeruli, which is in part due to exudation of inflammatory cells (neutrophils and macrophages) and in part due to proliferation of endogenous glomerular cells
Hypercellularity of glomeruli, which is in part due to exudation of inflammatory cells (neutrophils and macrophages) and in part due to proliferation of endogenous glomerular cells




30 Describe the most common cause of postinfectious glomerulonephritis
Group A beta-hemolytic streptococci (Streptococcus pyogenes) account for 90% of all glomerulonephritis cases. Glomerulonephritis typically occurs 1 to 4 weeks after a strep throat or skin infection (impetigo) caused by one of the nephritogenic strains of this microbe. Occasionally the same clinical and pathologic findings may follow staphylococcal infection and even some viral diseases, such as hepatitis B or C or hepatitis caused by human immunodeficiency virus (HIV). See Fig. 15-3.

33 Why is the concentration of serum complement C3 low in acute poststreptococcal glomerulonephritis?
34 What is the typical outcome and what are the possible long-term consequences of acute poststreptococcal glomerulonephritis?
The disease has a better prognosis in children than in adults:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


