Chapter 9 The Hematopoietic and Lymphoid Systems









RED BLOOD CELL DISORDERS






















4 List three major groups of anemia according to their etiology and pathogenesis



 Anemia due to increased rate of RBC destruction (hemolytic anemia)
Anemia due to increased rate of RBC destruction (hemolytic anemia)




5 How are anemias classified according to the red cell size and shape and their hemoglobin content?








TABLE 9-1 Abnormal Red Blood Cell (RBC) Morphology
| Abnormality | Features | Significance |
|---|---|---|
| Anisocytosis | Variation in RBC size | Nonspecific |
| Poikilocytosis | Variation in RBC shape | Nonspecific |
| Target cells | Targetlike appearance | Thalassemia, hemoglobinopathies |
| Sickle cells | Bipolar (sickle) or hollyleaf | Sickle cell anemia RBCs |
| Schistocytes | RBC fragments | Microangiopathic hemolytic anemia |
| Teardrops | Tennis racket RBCs | Myelofibrosis, severe anemias |
| Spherocytes | Spherical RBCs with dense hemoglobin content | Hereditary spherocytosis, alcoholism |
| Bite cells | Smooth semicircle taken from one edge | G6PD deficiency |
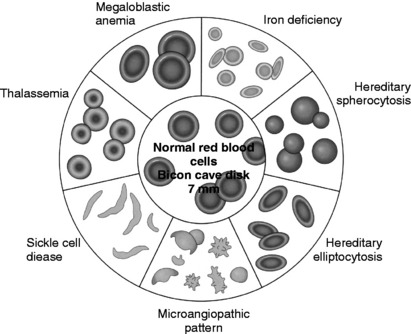
6 Define the main hematologic parameters

















7 Discuss how reticulocyte counts are used in clinical practice
 Hypoproliferative: Patients with anemia caused by defects in erythrocyte proliferation or maturation tend to have low reticulocyte counts. Patients suffering from pernicious anemia have low reticulocyte count, which will, however, increase after vitamin B12 treatment.
Hypoproliferative: Patients with anemia caused by defects in erythrocyte proliferation or maturation tend to have low reticulocyte counts. Patients suffering from pernicious anemia have low reticulocyte count, which will, however, increase after vitamin B12 treatment.







13 What is the difference between intravascular and extravascular hemolysis?
 Intravascular hemolysis: Significant lysis of erythrocytes rarely occurs within the vascular spaces. In intravascular hemolysis, normal erythrocytes are damaged by:
Intravascular hemolysis: Significant lysis of erythrocytes rarely occurs within the vascular spaces. In intravascular hemolysis, normal erythrocytes are damaged by:





14 What are the main features of intravascular hemolysis?
 Jaundice: It is related to the excessive formation of unconjugated bilirubin from the heme portion of hemoglobin. Unconjugated bilirubin is bound to albumin and does not appear in urine.
Jaundice: It is related to the excessive formation of unconjugated bilirubin from the heme portion of hemoglobin. Unconjugated bilirubin is bound to albumin and does not appear in urine.







17 What is the difference between anemia caused by extrinsic factors (extracorpuscular defects) and anemia caused by intrinsic factors (intracorpuscular defects)?



 Intracorpuscular
Intracorpuscular Hereditary disorders (i.e., red cell membrane disorders, red cell enzyme deficiencies, and disorders of hemoglobin synthesis)
Hereditary disorders (i.e., red cell membrane disorders, red cell enzyme deficiencies, and disorders of hemoglobin synthesis)
Key Points: Hematology and Red Blood Cell Disorders
19 What are the exact molecular defects in HS?
The normal membrane cytoskeleton of RBCs is composed of several proteins, the most important of which are α and β spectrin, ankyrin, actin, and proteins known as band 4.1 and band 3 (Fig. 9-2). Together these proteins maintain the normal biconcave shape of RBCs. The mutation of ankyrin gene is the most common defect in autosomal dominant HS, and the mutations of gene encoding protein band 3 account for 20% of cases. Genes encoding other cytoskeletal proteins are less often mutated.
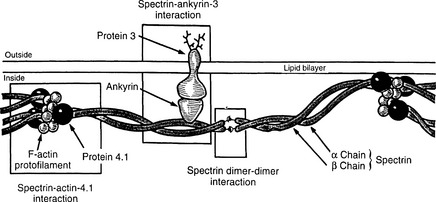
21 Describe the pathologic findings in HS
The most prominent changes are found in the peripheral blood, the bone marrow, and the spleen:
 Blood smears: Spheroid red cells appear uniformly red in routine smears (spherocytosis). Their central pallor, corresponding to the central concavity, is lacking. Although this morphology of RBCs is distinctive, it is not pathognomonic of HS and may also be seen in autoimmune hemolytic anemias.
Blood smears: Spheroid red cells appear uniformly red in routine smears (spherocytosis). Their central pallor, corresponding to the central concavity, is lacking. Although this morphology of RBCs is distinctive, it is not pathognomonic of HS and may also be seen in autoimmune hemolytic anemias.
 Spleen
Spleen Splenomegaly of moderate extent (500–1000 g) results from marked congestion of the red pulp cords. The sinuses appear virtually empty.
Splenomegaly of moderate extent (500–1000 g) results from marked congestion of the red pulp cords. The sinuses appear virtually empty.





23 What is the clinical course of hereditary spherocytosis?
The severity of the disease is variable among individuals:
 In a minority of patients, hereditary spherocytosis presents at birth with marked jaundice, requiring exchange transfusion.
In a minority of patients, hereditary spherocytosis presents at birth with marked jaundice, requiring exchange transfusion.

25 What should one know about G6PD deficiency?








28 Describe the difference between the sickle cell trait and sickle cell anemia
 When an individual is homozygous for the mutant gene, all the hemoglobin is of the abnormal HbS type. Sickling and hemolysis occur in regular living circumstances, causing the spectrum of clinical signs and symptoms known as sickle cell anemia.
When an individual is homozygous for the mutant gene, all the hemoglobin is of the abnormal HbS type. Sickling and hemolysis occur in regular living circumstances, causing the spectrum of clinical signs and symptoms known as sickle cell anemia.
29 Why do dehydration and anoxia (e.g., high altitude) potentiate sickling of RBCs in sickle cell anemia?
30 Name two main causes of ischemia in sickle cell anemia
 Chronic hemolytic anemia: Anemia results from shortened life span of RBC, which survive on average only 20 days in circulation. In response to chronic hemolysis the bone marrow undergoes massive hyperplasia (Fig. 9-3).
Chronic hemolytic anemia: Anemia results from shortened life span of RBC, which survive on average only 20 days in circulation. In response to chronic hemolysis the bone marrow undergoes massive hyperplasia (Fig. 9-3).
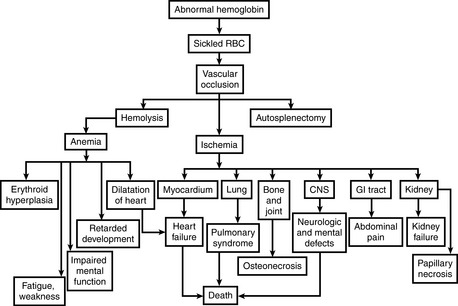
33 Discuss the most common bacteria causing infection, death, or both in children with sickle cell anemia
34 What are three types of crisis in sickle cell anemia?
 Vasoocclusive (or painful) crises: Episodes of hypoxia and infarction are associated with severe pain in the affected area. Most commonly involved sites are bones, lungs, liver, brain, spleen, and penis.
Vasoocclusive (or painful) crises: Episodes of hypoxia and infarction are associated with severe pain in the affected area. Most commonly involved sites are bones, lungs, liver, brain, spleen, and penis. In children, painful bone crises are extremely common, causing the so-called hand–foot syndrome, which can be distinguished from acute osteomyelitis with difficulty.
In children, painful bone crises are extremely common, causing the so-called hand–foot syndrome, which can be distinguished from acute osteomyelitis with difficulty.













38 What are the pathologic features of β-thalassemia?






 Bones
Bones Most erythroblasts die in the bone marrow (apoptosis), resulting in ineffective erythropoiesis. This, in turn, stimulates erythropoietin secretion, which leads to severe erythroid hyperplasia in the bone marrow and often at extramedullary sites.
Most erythroblasts die in the bone marrow (apoptosis), resulting in ineffective erythropoiesis. This, in turn, stimulates erythropoietin secretion, which leads to severe erythroid hyperplasia in the bone marrow and often at extramedullary sites.

 Spleen
Spleen Sequestration and destruction of inclusion-bearing red cells is derived from precursors that escape intramedullary death.
Sequestration and destruction of inclusion-bearing red cells is derived from precursors that escape intramedullary death.






40 What is the difference between the three clinical types of β-thalassemia (thalassemia major, intermedia, and minor)?
The clinical classification of β-thalassemias is based on:



 β-Thalassemia major
β-Thalassemia major Severe transfusion-dependent anemia that first becomes manifest 6 to 9 months after birth as hemoglobin synthesis switches from HbF to HbA
Severe transfusion-dependent anemia that first becomes manifest 6 to 9 months after birth as hemoglobin synthesis switches from HbF to HbA












42 List the four main clinical types of α-thalassemia
These are classified on the basis of the number and position of the α-globin genes deleted:


















46 What is the difference between warm antibody and cold antibody hemolytic anemia?






 Cold antibody hemolytic anemia:
Cold antibody hemolytic anemia: IgM antibodies bind to the RBC. Two distinct forms are recognized:
IgM antibodies bind to the RBC. Two distinct forms are recognized:






49 List the main hematologic features of megaloblastic anemia
Peripheral blood shows the following features:






50 What are the changes seen in bone marrow in megaloblastic anemia?
Bone marrow shows the following changes:














53 What is the pathogenesis of pernicious anemia?
 Ineffective erythropoiesis (megaloblasts are especially prone to intramedullary destruction; premature destruction of granulocytic and platelet precursors also occurs)
Ineffective erythropoiesis (megaloblasts are especially prone to intramedullary destruction; premature destruction of granulocytic and platelet precursors also occurs)Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


