48
The Extracellular Matrix
Robert K. Murray, MD, PhD & Frederick W. Keeley, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Appreciate the importance of the extracellular matrix (ECM) and its components in health and disease;
Appreciate the importance of the extracellular matrix (ECM) and its components in health and disease;
![]() Describe the structural and functional properties of collagen and elastin, the major proteins of the ECM;
Describe the structural and functional properties of collagen and elastin, the major proteins of the ECM;
![]() Indicate the major features of fibrillin, fibronectin, and laminin, other important proteins of the ECM;
Indicate the major features of fibrillin, fibronectin, and laminin, other important proteins of the ECM;
![]() Describe the properties and general features of the synthesis and degradation of glycosaminoglycans and proteoglycans, and their contributions to the ECM;
Describe the properties and general features of the synthesis and degradation of glycosaminoglycans and proteoglycans, and their contributions to the ECM;
![]() Give a brief account of the major biochemical features of bone and cartilage.
Give a brief account of the major biochemical features of bone and cartilage.
BIOMEDICAL IMPORTANCE
Most mammalian cells are located in tissues where they are surrounded by a complex ECM often referred to as “connective tissue.” The ECM contains three major classes of biomolecules: (1) structural proteins, for example, collagen, elastin, and fibrillin-1; (2) certain specialized proteins such as fibronectin and laminin; and (3) proteoglycans, whose chemical natures are described below. The ECM has been found to be involved in many normal and pathologic processes—for example, it plays important roles in development, in inflammatory states, and in the spread of cancer cells. Involvement of certain components of the ECM has been documented in both rheumatoid arthritis and osteoarthritis. Several diseases (eg, osteogenesis imperfecta and a number of types of the Ehlers-Danlos syndrome) are due to genetic disturbances of the synthesis of collagen. Specific components of proteoglycans (the glycosaminoglycans; GAGs) are affected in the group of genetic disorders known as the mucopolysaccharidoses. Changes occur in the ECM during the aging process. This chapter describes the basic biochemistry of the three major classes of biomolecules found in the ECM and illustrates their biomedical significance. Major biochemical features of two specialized forms of ECM—bone and cartilage—and of a number of diseases involving them are also briefly considered.
COLLAGEN TYPE I IS COMPOSED OF A TRIPLE HELIX STRUCTURE & FORMS FIBRILS
Collagen, the major component of most connective tissues, constitutes approximately 25% of the protein of mammals. It provides an extracellular framework for all metazoan animals and exists in virtually every animal tissue. At least 28 distinct types of collagen made up of over 30 distinct polypeptide chains (each encoded by a separate gene) have been identified in human tissues. Although several of these are present only in small proportions, they may play important roles in determining the physical properties of specific tissues. In addition, a number of proteins (eg, the C1q component of the complement system, pulmonary surfactant proteins SPA and SPD) that are not classified as collagens have collagen-like domains in their structures; these proteins are sometimes referred to as “noncollagen collagens.”
Table 48-1 summarizes information on many of the types of collagens found in human tissues; the nomenclature used to designate types of collagen and their genes is described in the footnote.
TABLE 48–1 Types of Collagen and Their Genes1
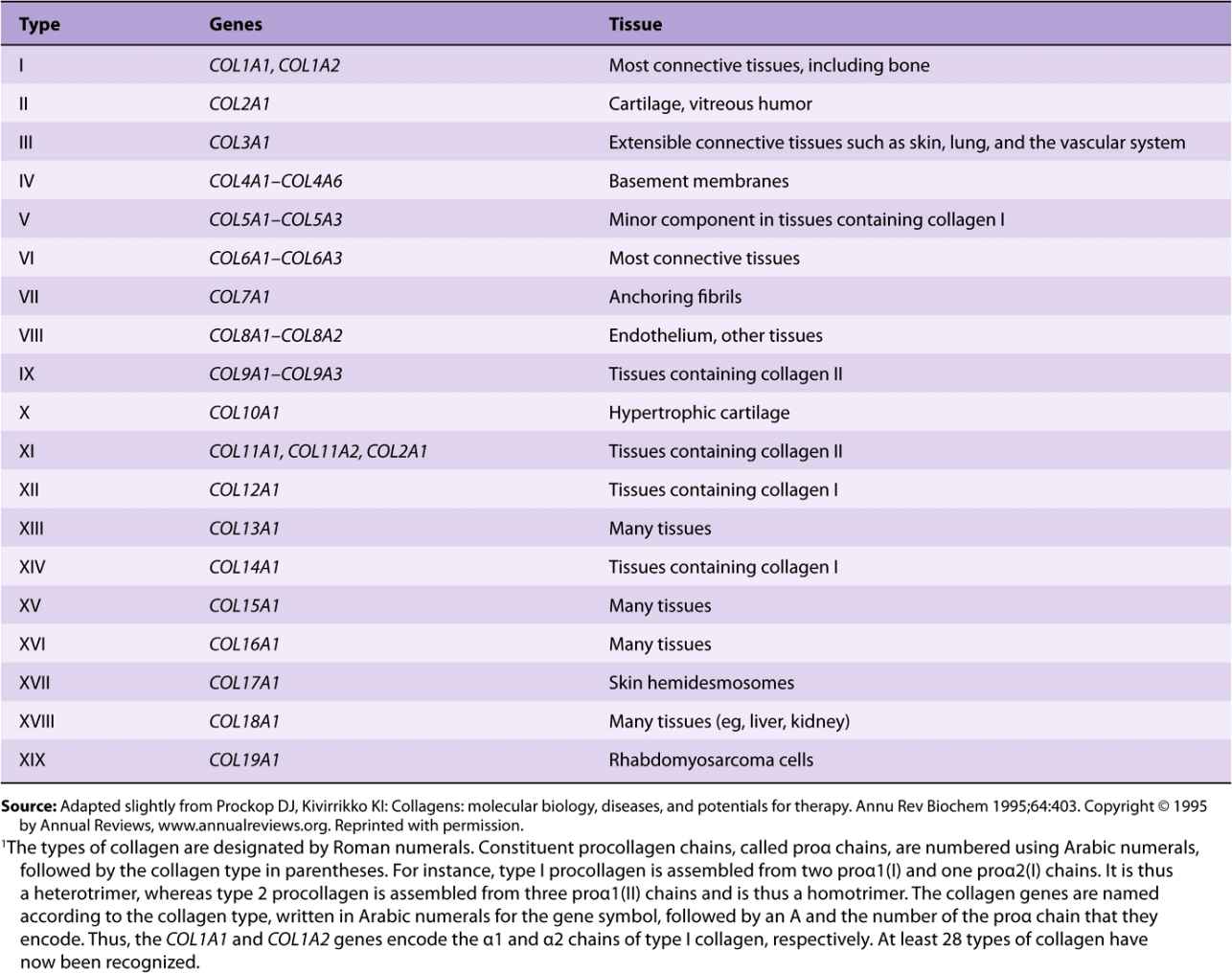
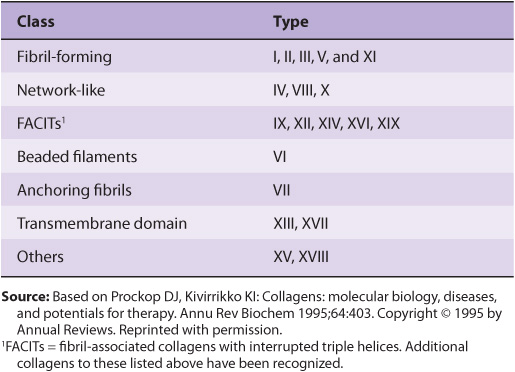
In Table 48-2, the types of collagen listed in Table 48-1 are subdivided into a number of classes based primarily on the structures they form. In this chapter, we shall be primarily concerned with the fibril-forming collagens I and II, the major collagens of skin and bone and of cartilage, respectively. However, mention will be made of some of the other collagens.
TABLE 48–2 Classification of Collagens, Based Primarily on the Structures That They Form
COLLAGEN IS THE MOST ABUNDANT PROTEIN IN THE ANIMAL WORLD
All collagen types have a triple helical structure. In some collagens, the entire molecule is triple helical, whereas in others the triple helix may involve only a fraction of the structure. Mature collagen type I, containing approximately 1000 amino acids, belongs to the former type; in it, each polypeptide subunit or alpha chain is twisted into a left-handed polyproline helix of three residues per turn (Figure 48–1). Three of these alpha chains are then wound into a right-handed superhelix, forming a rodlike molecule 1.4 nm in diameter and about 300 nm long. A striking characteristic of collagen is the occurrence of glycine residues at every third position of the triple helical portion of the alpha chain. This is necessary because glycine is the only amino acid small enough to be accommodated in the limited space available down the central core of the triple helix. This repeating structure, represented as (Gly-X-Y)n, is an absolute requirement for the formation of the triple helix. While X and Y can be any other amino acids, about 100 of the X positions are proline and about 100 of the Y positions are hydroxyproline. Proline and hydroxyproline confer rigidity on the collagen molecule. Hydroxyproline is formed by the posttranslational hydroxylation of peptide-bound proline residues catalyzed by the enzyme prolyl hydroxylase, whose cofactors are ascorbic acid (vitamin C) and α-ketoglutarate. Lysines in the Y position may also be posttranslationally modified to hydroxylysine through the action of lysyl hydroxylase, an enzyme with similar cofactors. Some of these hydroxylysines may be further modified by the addition of galactose or galactosyl-glucose through an O-glycosidic linkage (see Chapter 47), a glycosylation site that is unique to collagen.
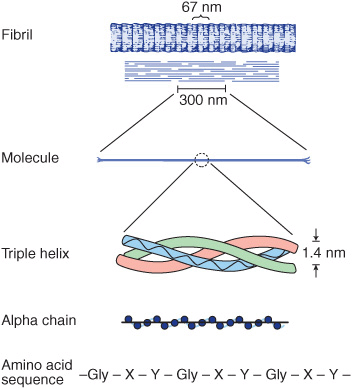
FIGURE 48–1 Molecular features of the collagen structure from primary sequence up to the fibril. Each individual polypeptide chain is twisted into a left-handed helix of three residues (Gly-X-Y) per turn, and all of these chains are then wound into a right-handed superhelix. (Slightly modified and reproduced, with permission, from Eyre DR: Collagen (1980), “Molecular diversity in the body’s protein scaffold”. Science 207:1315. Reprinted with permission from AAAS.)
Collagen types that form long rod-like fibers in tissues are assembled by lateral association of these triple helical units into a “quarter staggered” alignment such that each is displaced longitudinally from its neighbor by slightly less than one-quarter of its length (Figure 48–1, upper part). This arrangement is responsible for the banded appearance of these fibers in connective tissues. Collagen fibers are further stabilized by the formation of covalent cross-links, both within and between the triple helical units. These cross-links form through the action of lysyl oxidase, a copper-dependent enzyme that oxidatively deaminates the ε-amino groups of certain lysine and hydroxylysine residues, yielding reactive aldehydes. Such aldehydes can form aldol condensation products with other lysine- or hydroxylysine-derived aldehydes or form Schiff bases with the ε-amino groups of unoxidized lysines or hydroxylysines. These reactions, after further chemical rearrangements, result in the stable covalent cross-links that are important for the tensile strength of the fibers. Histidine may also be involved in certain cross-links.
Several collagen types do not form fibrils in tissues (Table 48-2). They are characterized by interruptions of the triple helix with stretches of protein lacking Gly-X-Y repeat sequences. These non-Gly-X-Y sequences result in areas of globular structure interspersed in the triple helical structure.
Type IV collagen, the best-characterized example of a collagen with discontinuous triple helices, is an important component of basement membranes, where it forms a meshlike network.
Collagen Undergoes Extensive Posttranslational Modifications
Newly synthesized collagen undergoes extensive posttranslational modification before becoming part of a mature extracellular collagen fiber (Table 48-3). Like most secreted proteins, collagen is synthesized on ribosomes in a precursor form, preprocollagen, which contains a leader or signal sequence that directs the polypeptide chain into the lumen of the endoplasmic reticulum. As it enters the endoplasmic reticulum, this leader sequence is enzymatically removed. Hydroxylation of proline and lysine residues and glycosylation of hydroxylysines in the procollagen molecule also take place at this site. The procollagen molecule contains polypeptide extensions (extension peptides) of 20-35 kDa at both its amino and carboxyl terminal ends, neither of which is present in mature collagen. Both extension peptides contain cysteine residues. While the amino terminal propeptide forms only intrachain disulfide bonds, the carboxyl terminal propeptides form both intrachain and interchain disulfide bonds. Formation of these disulfide bonds assists in the registration of the three collagen molecules to form the triple helix, winding from the carboxyl terminal end. After formation of the triple helix, no further hydroxylation of proline or lysine or glycosylation of hydroxylysines can take place. Self-assembly is a cardinal principle in the biosynthesis of collagen.
TABLE 48–3 Order and Location of Processing of the Fibrillar Collagen Precursor

Following secretion from the cell by way of the Golgi apparatus, extracellular enzymes called procollagen aminoproteinase and procollagen carboxyproteinase remove the extension peptides at the amino and carboxyl terminal ends, respectively. Cleavage of these propeptides may occur within crypts or folds in the cell membrane. Once the propeptides are removed, the triple helical collagen molecules, containing approximately 1000 amino acids per chain, spontaneously assemble into collagen fibers. These are further stabilized by the formation of inter- and intrachain cross-links through the action of lysyl oxidase, as described previously.
The same cells that secrete collagen also secrete fibronectin, a large glycoprotein present on cell surfaces, in the extracellular matrix, and in blood (see below). Fibronectin binds to aggregating precollagen fibers and alters the kinetics of fiber formation in the pericellular matrix. Associated with fibronectin and procollagen in this matrix are the proteoglycans heparan sulfate and chondroitin sulfate (see below). In fact, type IX collagen, a minor collagen type from cartilage, contains an attached glycosaminoglycan chain. Such interactions may serve to regulate the formation of collagen fibers and to determine their orientation in tissues.
Once formed, collagen is relatively metabolically stable. However, its breakdown is increased during starvation and various inflammatory states. Excessive production of collagen occurs in a number of conditions, for example, hepatic cirrhosis.
A Number of Genetic Diseases Result from Abnormalities in the Synthesis of Collagen
About 30 genes encode the collagens, and their pathway of biosynthesis is complex, involving at least eight enzyme-catalyzed posttranslational steps. Thus, it is not surprising that a number of diseases (Table 48-4) are due to mutations in collagen genes or in genes encoding some of the enzymes involved in these post-translational modifications. The diseases affecting bone (eg, osteogenesis imperfecta) and cartilage (eg, the chondrodysplasias) will be discussed later in this chapter.
TABLE 48–4 Diseases Caused by Mutations in Collagen Genes or by Deficiencies in the Activities of Posttranslational Enzymes Involved in the Biosynthesis of Collagen
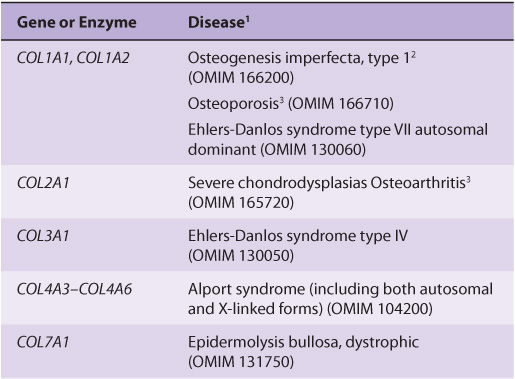

The Ehlers-Danlos syndrome comprises a group of inherited disorders whose principal clinical features are hyperextensibility of the skin, abnormal tissue fragility, and increased joint mobility. The clinical picture is variable, reflecting underlying extensive genetic heterogeneity. At least 10 types have been recognized, most but not all of which reflect a variety of lesions in the synthesis of collagen. Type IV is the most serious because of its tendency for spontaneous rupture of arteries or the bowel, reflecting abnormalities in type III collagen. Patients with type VI, due to a deficiency of lysyl hydroxylase, exhibit marked joint hypermobility and a tendency to ocular rupture. A deficiency of procollagen N-proteinase, causing formation of abnormal thin, irregular collagen fibrils, results in type VIIC, manifested by marked joint hypermobility and soft skin.
The Alport syndrome is the designation applied to a number of genetic disorders (both X-linked and autosomal) affecting the structure of type IV collagen fibers, the major collagen found in the basement membranes of the renal glomeruli (see discussion of laminin, in the following). Mutations in several genes encoding type IV collagen fibers have been demonstrated. The presenting sign is hematuria, and patients may eventually develop end-stage renal disease. Electron microscopy reveals characteristic abnormalities of the structure of the basement membrane and lamina densa.
In epidermolysis bullosa, the skin breaks and blisters as a result of minor trauma. The dystrophic form is due to mutations in COL7A1, affecting the structure of type VII collagen. This collagen forms delicate fibrils that anchor the basal lamina to collagen fibrils in the dermis. These anchoring fibrils have been shown to be markedly reduced in this form of the disease, probably resulting in the blistering. Epidermolysis bullosa simplex, another variant, is due to mutations in keratin 5 (Chapter 49).
Scurvy affects the structure of collagen. However, it is due to a deficiency of ascorbic acid (Chapter 44), and is not a genetic disease. Its major signs are bleeding gums, subcutaneous hemorrhages, and poor wound healing. These signs reflect impaired synthesis of collagen due to deficiencies of prolyl and lysyl hydroxylases, both of which require ascorbic acid as a cofactor.
In Menkes disease deficiency of copper results in defective cross-linking of collagen and elastin by the copper-dependent enzyme lysyl oxidase. (Menkes disease is discussed in Chapter 50.)
ELASTIN CONFERS EXTENSIBILITY & RECOIL ON LUNG, BLOOD VESSELS & LIGAMENTS
Elastin is a connective tissue protein that is responsible for properties of extensibility and elastic recoil in tissues. Although not as widespread as collagen, elastin is present in large amounts, particularly in tissues that require these physical properties, for example, lung, large arterial blood vessels, and some elastic ligaments. Smaller quantities of elastin are also found in skin, ear cartilage, and several other tissues. In contrast to collagen, there appears to be only one genetic type of elastin, although variants arise by alternative splicing (Chapter 36) of the hnRNA for elastin. Elastin is synthesized as a soluble monomer of ~70 kDa called tropoelastin. Some of the prolines of tropoelastin are hydroxylated to hydroxyproline by prolyl hydroxylase, though hydroxylysine and glycosylated hydroxylysine are not present. Unlike collagen, tropoelastin is not synthesized in a pro-form with extension peptides. Furthermore, elastin does not contain repeat Gly-X-Y sequences, triple helical structure, or carbohydrate moieties.
After secretion from the cell, certain lysyl residues of tropoelastin are oxidatively deaminated to aldehydes by lysyl oxidase, the same enzyme involved in this process in collagen. However, the major cross-links formed in elastin are the desmosines, which result from the condensation of three of these lysine-derived aldehydes with an unmodified lysine to form a tetrafunctional cross-link unique to elastin. Once cross-linked in its mature, extracellular form, elastin is highly insoluble and extremely stable and has a very low-turnover rate. Elastin exhibits a variety of random coil conformations that permit the protein to stretch and subsequently recoil during the performance of its physiologic functions.
Table 48-5 summarizes the main differences between collagen and elastin.
TABLE 48–5 Major Differences Between Collagen and Elastin
Deletions in the elastin gene (located at 7q11.23) have been found in approximately 90% of subjects with the Williams-Beuren syndrome (OMIM 194050), a developmental disorder affecting connective tissue and the central nervous system. The mutations, by affecting synthesis of elastin, probably play a causative role in the supravalvular aortic stenosis often found in this condition. Fragmentation or, alternatively, a decrease of elastin is found in conditions such as pulmonary emphysema, cutis laxa, and aging of the skin.
MARFAN SYNDROME IS DUE TO MUTATIONS IN THE GENE FOR FIBRILLIN-1, A PROTEIN PRESENT IN MICROFIBRILS
The Marfan syndrome is a relatively prevalent inherited disease affecting connective tissue; it is inherited as an autosomal dominant trait. It affects the eyes (eg, causing dislocation of the lens, known as ectopia lentis), the skeletal system (most patients are tall and exhibit long digits [arachnodactyly] and hyperextensibility of the joints), and the cardiovascular system (eg, causing weakness of the aortic media, leading to dilation of the ascending aorta). Abraham Lincoln may have had this condition. Most cases are caused by mutations in the gene (on chromosome 15) for fibrillin-1; missense mutations have been detected in several patients with the Marfan syndrome. This results in abnormal fibrillin and/or lower amounts being deposited in the ECM. There is evidence that the cytokine TGF-β normally binds to fibrillin-1, and if this binding is decreased (due to lower amounts of fibrillin-1), this can lead to an excess of the cytokine. The excess of TGF-β may contribute to the pathology (eg, in the aorta and aortic valve) found in the syndrome. This finding may lead to the development of therapies for the condition using drugs that antagonize TGF-β (eg, Losartan).
Fibrillin-1 is a large glycoprotein (about 350 kDa) that is a structural component of microfibrils, 10- to 12-nm fibers found in many tissues. It is secreted (subsequent to a proteolytic cleavage) into the ECM by fibroblasts and becomes incorporated into the insoluble microfibrils, which appear to provide a scaffold for deposition of elastin. Of special relevance to the Marfan syndrome, fibrillin-1 is found in the zonular fibers of the lens, in the periosteum, and associated with elastin fibers in the aorta (and elsewhere); these locations respectively explain the ectopia lentis, arachnodactyly, and cardiovascular problems found in the syndrome. Other proteins (eg, emelin and two microfibril-associated proteins) are also present in microfibrils. It appears likely that their abnormalities may cause other connective tissue disorders. A gene for another fibrillin—fibrillin-2—exists on chromosome 5; mutations in this gene are linked to causation of congenital contractural arachnodactyly (OMIM 121050), but not to the Marfan syndrome. Fibrillin-2 may be important in deposition of microfibrils early in development. The probable sequence of events leading to Marfan syndrome is summarized in Figure 48–2.

FIGURE 48–2 Probable sequence of events in the causation of the major signs exhibited by patients with the Marfan syndrome (OMIM 154700).
FIBRONECTIN IS AN IMPORTANT GLYCOPROTEIN INVOLVED IN CELL ADHESION & MIGRATION
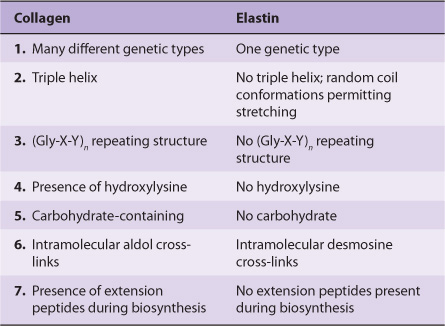
Fibronectin is a major glycoprotein of the extracellular matrix, also found in a soluble form in plasma. It consists of two identical subunits, each of about 230 kDa, joined by two disulfide bridges near their carboxyl terminals. The gene encoding fibronectin is very large, containing some 50 exons; the RNA produced by its transcription is subject to considerable alternative splicing, and as many as 20 different mRNAs have been detected in various tissues. Fibronectin contains three types of repeating motifs (I, II, and III), which are organized into functional domains (at least seven); functions of these domains include binding heparin (see below) and fibrin, collagen, DNA, and cell surfaces (Figure 48–3). The amino acid sequence of the fibronectin receptor of fibroblasts has been derived, and the protein is a member of the transmembrane integrin class of proteins (Chapter 51). The integrins are heterodimers, containing various types of α and β polypeptide chains. Fibronectin contains an Arg-Gly-Asp (RGD) sequence that binds to the receptor. The RGD sequence is shared by a number of other proteins present in the ECM that bind to integrins present in cell surfaces. Synthetic peptides containing the RGD sequence inhibit the binding of fibronectin to cell surfaces. Figure 48–4 illustrates the interaction of collagen, fibronectin, and laminin, all major proteins of the ECM, with a typical cell (eg, fibroblast) present in the matrix.
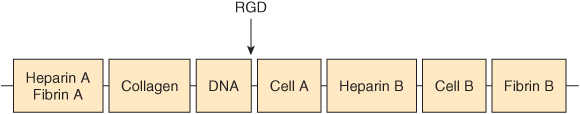
FIGURE 48–3 Schematic representation of fibronectin. Seven functional domains of fibronectin are represented; two differenttypesof domain for heparin, cell binding, and fibrin are shown. The domains are composed of various combinations of three structural motifs (I, II, and III), not depicted in the figure. Also not shown is the fact that fibronectin is a dimer joined by disulfide bridges near the carboxyl terminals of the monomers. The approximate location of the RGD sequence of fibronectin, which interacts with a variety of fibronectin integrin receptors on cell surfaces, is indicated by the arrow. (Redrawn after Yamada KM: Adhesive recognition sequences. J Biol Chem 1991;266:12809.)
FIGURE 48–4 Schematic representation of a cell interacting through various integrin receptors with collagen, fibronectin, and laminin present in the ECM. (Specific subunits are not indicated.) (Redrawn after Yamada KM: Adhesive recognition sequences. J Biol Chem 1991;266:12809.)
The fibronectin receptor interacts indirectly with actin microfilaments (Chapter 49) present in the cytosol (Figure 48–5). A number of proteins, collectively known as attachment proteins
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


