18
The Eicosanoids: Prostaglandins, Thromboxanes, Leukotrienes, & Related Compounds
The eicosanoids are oxygenation products of polyunsaturated long-chain fatty acids. They are ubiquitous in the animal kingdom and are also found—together with their precursors—in a variety of plants. They constitute a very large family of compounds that are highly potent and display an extraordinarily wide spectrum of biologic activity. Because of their biologic activity, the eicosanoids, their specific receptor antagonists and enzyme inhibitors, and their plant and fish oil precursors have great therapeutic potential.
ARACHIDONIC ACID & OTHER POLYUNSATURATED PRECURSORS
Arachidonic acid (AA), or 5,8,11,14-eicosatetraenoic acid, the most abundant of the eicosanoid precursors, is a 20-carbon (C20) fatty acid containing four double bonds (designated C20:4–6). The first double bond in AA occurs at 6 carbons from the methyl end, defining AA as an omega-6 fatty acid. AA must first be released or mobilized from the sn-2 position of membrane phospholipids by one or more lipases of the phospholipase A2 (PLA2) type (Figure 18–1) for eicosanoid synthesis to occur. The phospholipase A2 superfamily consists of 15 groups, with at least three classes of phospholipases contributing to arachidonate release from membrane lipids: (1) cytosolic (c) PLA2, and (2) secretory (s) PLA2, which are calcium-dependent; and (3) calcium-independent (i) PLA2. Chemical and physical stimuli activate the Ca2+-dependent translocation of cPLA2, which has high affinity for AA, to the membrane, where it releases arachidonate. Arachidonate release via iPLA2 and various sPLA2 subtypes also has been examined. Under nonstimulated conditions, AA liberated by iPLA2 is reincorporated into cell membranes, so there is negligible eicosanoid biosynthesis. While cPLA2 dominates in the acute release of AA, inducible sPLA2 contributes under conditions of sustained or intense stimulation of AA production. AA can also be released from phospholipase C-generated diacylglcerol esters by the action of diacylglycerol and monoacylglycerol lipases.
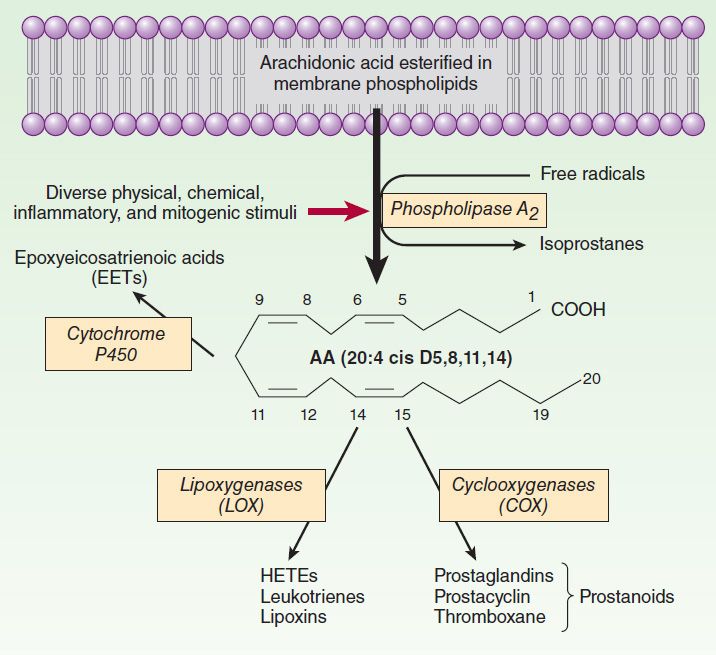
FIGURE 18–1 Pathways of arachidonic acid (AA) release and metabolism.
Following mobilization, AA is oxygenated by four separate routes: enzymatically via the cyclooxygenase (COX), lipoxygenase, and P450 epoxygenase pathways; and nonenzymatically via the isoeicosanoid pathway (Figure 18–1). Among factors determining the type of eicosanoid synthesized are (1) the substrate lipid species, (2) the type of cell, and (3) the manner in which the cell is stimulated. Distinct but related products can be formed from precursors other than AA. For example, homo-γ-linoleic acid (C20:3–6), also an omega-6 fatty acid, or eicosapentaenoic acid (C20:5–3), an omega-3 fatty acid, yield products that differ quantitatively and qualitatively from those derived from AA. This shift in product formation is the basis for dietary manipulation of eicosanoid generation using fatty acids obtained from cold-water fish or from plants as nutritional supplements in humans. For example, thromboxane (TXA2), a powerful vasoconstrictor and platelet agonist, is synthesized from AA via the COX pathway. COX metabolism of eicosapentaenoic acid yields TXA3, which is relatively inactive. 3-Series prostaglandins, such as prostaglandin E3 (PGE3), can also act as partial agonists or antagonists thereby reducing the activity of their AA-derived 2-series counterparts. The hypothesis that dietary eicosapentaenoate substitution for arachidonate could reduce the incidence of cardiovascular disease and cancer remains controversial.
ACRONYMS
SYNTHESIS OF EICOSANOIDS
Products of Prostaglandin Endoperoxide Synthases (Cyclooxygenases)
Two unique COX isozymes convert AA into prostaglandin endoperoxides. PGH synthase-1 (COX-1) is expressed constitutively in most cells. In contrast, PGH synthase-2 (COX-2) is more readily inducible; its expression varies depending on the stimulus. COX-2 is an immediate early-response gene product that is markedly up-regulated by shear stress, growth factors, tumor promoters, and cytokines, consistent with the presence of multiple regulatory motifs in the promoter and 3′ untranslated regions of the COX-2 gene. COX-1 generates prostanoids for “housekeeping” functions, such as gastric epithelial cytoprotection, whereas COX-2 is the major source of prostanoids in inflammation and cancer. This distinction is overly simplistic, however; there are both physiologic and pathophysiologic processes in which each enzyme is uniquely involved and others in which they function coordinately. For example, endothelial COX-2 is the primary source of vascular prostacyclin (PGI2), whereas renal COX-2-derived prostanoids are important for normal renal development and maintenance of function. Nonsteroidal anti-inflammatory drugs (NSAIDs; see Chapter 36) exert their therapeutic effects through inhibition of the COXs. Most older NSAIDs, like indomethacin, sulindac, meclofenamate, and ibuprofen nonselectively inhibit both COX-1 and COX-2, whereas the selective COX-2 inhibitors follow the order celecoxib = diclofenac = meloxicam = etodolac < valdecoxib << rofecoxib < lumiracoxib = etoricoxib for increasing COX-2 selectivity. Aspirin acetylates and inhibits both enzymes covalently and hence irreversibly. Low doses (< 100 mg/d) inhibit preferentially, but not exclusively, platelet COX-1, whereas higher doses inhibit both systemic COX-1 and COX-2. Genetic variations in human COX-2 variants have been linked with increased coronary heart disease risk, increases in some cancers, and reduced pain perception.
Both COX-1 and COX-2 function as homodimers inserted into the membrane of the endoplasmic reticulum to promote the uptake of two molecules of oxygen by cyclization of AA to yield a C9–C11 endoperoxide C15 hydroperoxide (Figure 18–2). This product is PGG2, which is then rapidly modified by the peroxidase moiety of the COX enzyme to add a 15-hydroxyl group that is essential for biologic activity. This product is PGH2. Both endoperoxides are highly unstable. Analogous families—PGH1 and PGH3 and their subsequent 1-series and 3-series products—are derived from homo-γ-linolenic acid and eicosapentaenoic acid, respectively. In both COX-1 and COX-2 homodimers, one protomer acts as the catalytic unit binding AA for oxygenation, while the other acts as an allosteric modifier of catalytic activity.
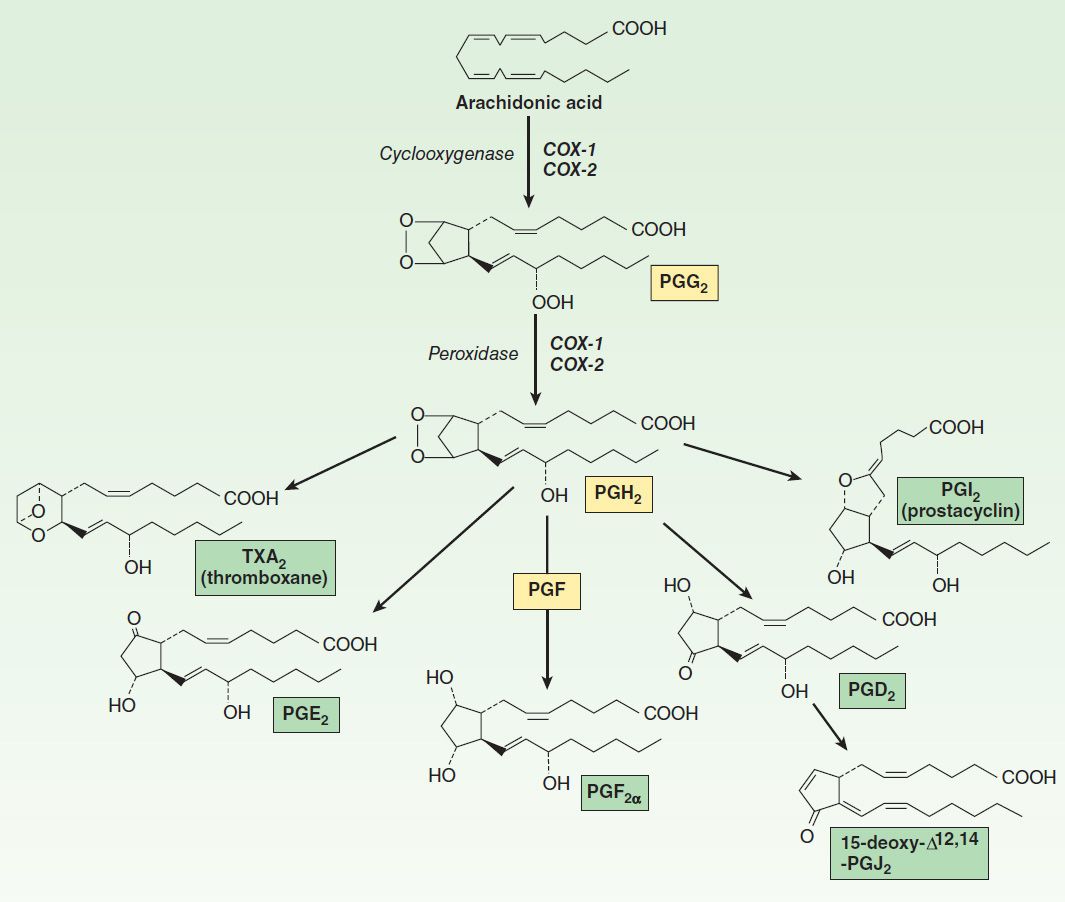
FIGURE 18–2 Prostanoid biosynthesis. Compound names are enclosed in boxes.
The prostaglandins, thromboxane, and prostacyclin, collectively termed the prostanoids, are generated from PGH2 through the action of downstream isomerases and synthases. These terminal enzymes are expressed in a relatively cell-specific fashion, such that most cells make one or two dominant prostanoids. The prostaglandins differ from each other in two ways: (1) in the substituents of the pentane ring (indicated by the last letter, eg, E and F in PGE and PGF) and (2) in the number of double bonds in the side chains (indicated by the subscript, eg, PGE1, PGE2). PGH2 is metabolized by prostacyclin, thromboxane, and PGF synthases (PGIS, TXAS, and PGFS) to PGI2, TXA2, and PGF2α, respectively. Two additional enzymes, 9,11-endoperoxide reductase and 9-ketoreductase, provide for PGF2α synthesis from PGH2 and PGE2, respectively. At least three PGE2 synthases have been identified: microsomal (m) PGES-1, the more readily inducible mPGES-2, and cytosolic PGES. There are two distinct PGDS isoforms, the lipocalin-type PGDS and the hematopoietic PGDS.
Several products of the arachidonate series are of current clinical importance. Alprostadil (PGE1) may be used for its smooth muscle relaxing effects to maintain the ductus arteriosus patent in some neonates awaiting cardiac surgery and in the treatment of impotence. Misoprostol, a PGE1 derivative, is a cytoprotective prostaglandin used in preventing peptic ulcer and in combination with mifepristone (RU-486) for terminating early pregnancies. Dinoprostone (PGE2) and PGF2α are used in obstetrics to induce labor. Latanoprost and several similar compounds are topically active PGF2α derivatives used in ophthalmology to reduce intraocular pressure in open-angle glaucoma or ocular hypertension. Prostacyclin (PGI2) is synthesized mainly by the vascular endothelium and is a powerful vasodilator and inhibitor of platelet aggregation. Synthetic PGI2 (epoprostenol) and PGI2 analogs (iloprost, treprostinil) are used to treat pulmonary hypertension and portopulmonary hypertension. In contrast, thromboxane (TXA2) has undesirable properties (aggregation of platelets, vasoconstriction). Therefore TXA2-receptor antagonists and synthesis inhibitors have been developed for cardiovascular indications, although these (except for aspirin) have yet to establish a place in clinical usage, and, in a recent large clinical trial, TXA2 receptor antagonism failed to show superiority over low-dose aspirin for secondary stroke protection.
All the naturally occurring COX products undergo rapid metabolism to inactive products either by hydration (for PGI2 and TXA2) or by oxidation of the key 15-hydroxyl group to the corresponding ketone by prostaglandin 15-hydroxy prostaglandin dehydrogenase (15-PGDH) after cellular uptake via an organic anion transporter polypeptide (OATP 2A1). Further metabolism is by δ13 reduction, β-oxidation, and ω-oxidation. The inactive metabolites are chemically stable and can be quantified in blood and urine by immunoassay or mass spectrometry as a measure of the in vivo synthesis of their parent compounds.
Products of Lipoxygenase
The metabolism of AA by the 5-, 12-, and 15-lipoxygenases (LOX) results in production of hydroperoxyeicosatetraenoic acids (HPETEs), which rapidly convert to hydroxy derivatives (HETEs). 5-LOX, the most actively investigated pathway, gives rise to the leukotrienes (Figure 18–3) and is present in leukocytes (neutrophils, basophils, eosinophils, and monocyte-macrophages) and other inflammatory cells such as mast cells and dendritic cells. This pathway is of great interest since it is associated with asthma, anaphylactic shock, and cardiovascular disease. Stimulation of these cells elevates intracellular Ca2+ and releases arachidonate; incorporation of molecular oxygen by 5-LOX, in association with 5-LOX-activating protein (FLAP), yields 5(S)-HPETE, which is then further converted by 5-LOX to the unstable epoxide leukotriene A4 (LTA4). This intermediate is either converted to the dihydroxy leukotriene B4 (LTB4), via the action of LTA4 hydrolase, or is conjugated with glutathione to yield leukotriene C4 (LTC4), by LTC4 synthase. Sequential degradation of the glutathione moiety by peptidases yields LTD4 and LTE4. These three products, LTC4, D4, and E4, are called cysteinyl leukotrienes. Although leukotrienes are predominantly generated in leukocytes, nonleukocyte cells (eg, endothelial cells) that express enzymes downstream of 5-LOX/FLAP can take up and convert leukocyte-derived LTA4 in a process termed transcellular biosynthesis. Transcellular formation of prostaglandins has also been shown; for example, endothelial cells can use platelet PGH2 to form PGI2.
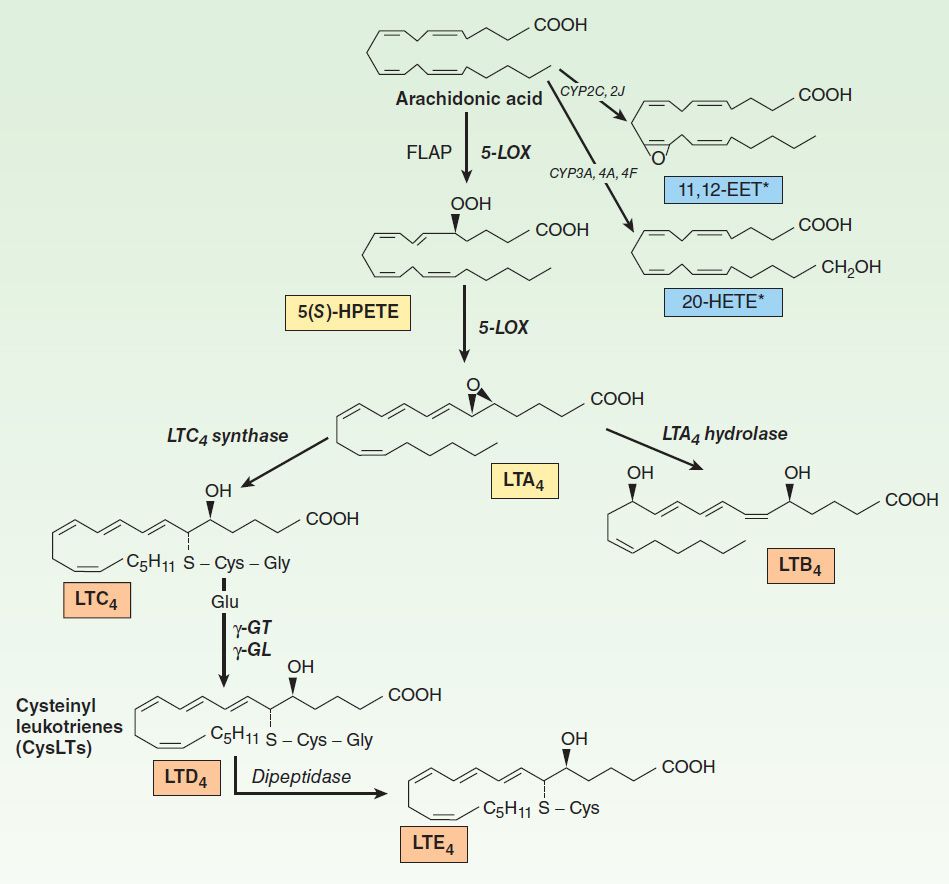
FIGURE 18–3 Leukotriene (LT) biosynthesis. LTC4, LTD4, and LTE4 are known collectively as the cysteinyl (Cys) LTs. FLAP, 5-LOX-activating protein; GT, glutamyl transpeptidase; GL, glutamyl leukotrienase. *Additional products include 5,6-; 8,9-; and 14,15-EET; and 19-, 18-, 17-, and 16-HETE.
LTC4 and LTD4 are potent bronchoconstrictors and are recognized as the primary components of the slow-reacting substance of anaphylaxis (SRS-A) that is secreted in asthma and anaphylaxis. There are four current approaches to antileukotriene drug development: 5-LOX enzyme inhibitors, cysteinyl leukotriene-receptor antagonists, inhibitors of FLAP, and phospholipase A2 inhibitors. Variants in the human 5-LOX gene (ALOX5) or the cysteinyl receptors (CYSLTR1 or CYSLTR2) have been linked with asthma and with the response to antileukotriene drugs.
LTA4, the primary product of 5-LOX, can be converted with appropriate stimulation via 12-LOX in platelets in vitro to the lipoxins LXA4 and LXB4 in vitro. These mediators can also be generated through 5-LOX metabolism of 15(S)-HETE, the product of 15-LOX-2 metabolism of arachidonic acid. The stereochemical isomer, 15(R)-HETE, may be derived from the action of aspirin-acetylated COX-2 and further transformed in leukocytes by 5-LOX to 15-epi-LXA4 or 15-epi-LXB4, the so-called aspirin-triggered lipoxins. The 15-LOX-1 isoform prefers linoleic acid as a substrate, forming 13(S)-hydroxyoctadecadienoic acid, while the sequential action of 15-LOX-1 and 5-LOX can convert the omega-3 fatty acid docosahexaenoic acid (DHA) to the resolvins, potentially anti-inflammatory, pro-resolving lipids. Synthetic resolvins, lipoxins, and epi-lipoxins exert anti-inflammatory actions when applied in vivo. Although these compounds can be formed from endogenous substrates in vitro and when synthesized may have potent biologic effects, the importance of the endogenous compounds in vivo in human biology remains ill defined. 12-HETE, a product of 12-LOX, can also undergo a catalyzed molecular rearrangement to epoxyhydroxyeicosatrienoic acids called hepoxilins. Proinflammatory effects of synthetic hepoxilins have been reported although their biologic relevance is unclear.
The epidermal LOXs, 12(R)-LOX and LOX-3, are distinct from “conventional” enzymes both in their natural substrates, which appear to not be arachidonic acid and linoleic acid, and in the products formed. Mutations in the genes for 12(R)-LOX (ALOX12B) or LOX-3 (ALOXE3) are commonly associated with autosomal recessive congenital ichthyosis, and epidermal accumulation of 12(R)-HETE is a feature of psoriasis and ichthyosis. Inhibitors of 12(R)-LOX are under investigation for the treatment of these proliferative skin disorders.
Epoxygenase Products
Specific isozymes of microsomal cytochrome P450 monooxygenases convert AA to hydroxy- or epoxyeicosatrienoic acids (Figures 18–1 and 18–3). The products are 20-HETE, generated by the CYP hydroxylases (CYP3A, 4A, 4F) and the 5,6-, 8,9-, 11,12-, and 14,15-epoxyeicosatrienoic acids (EETs), which arise from the CYP epoxygenase (2J, 2C). Their biosynthesis can be altered by pharmacologic, nutritional, and genetic factors that affect P450 expression. The biologic actions of the EETs are reduced by their conversion to the corresponding, and biologically less active, dihydroxyeicosatrienoic acids (DHETs) through the action of soluble epoxide hydrolase (sEH). Unlike the prostaglandins, the EETs can be esterified into phospholipids, which then act as storage sites. Intracellular fatty acid-binding proteins promote EET uptake into cells, incorporation into phospholipids, and availability to sEH. EETs are synthesized in endothelial cells and cause vasodilation in a number of vascular beds by activating the smooth muscle large conductance Ca2+-activated K+ channels. This results in smooth muscle cell hyperpolarization and vasodilation, leading to reduced blood pressure. Substantial evidence indicates that EETs may function as endothelium-derived hyperpolarizing factors, particularly in the coronary circulation. 15(S)-Hydroxy-11,12-EET, which arises from the 15-LOX pathway, is also an endothelium-derived hyperpolarizing factor and a substrate for sEH. Consequently there is interest in inhibitors of soluble sEH as potential antithrombotic and antihypertensive drugs. An exception to the general response to EETs as vasodilators is the pulmonary vasculature where they cause vasoconstriction. It is unclear yet whether this activity of EETs may limit the potential clinical use of sEH inhibitors. Down-regulation of pulmonary sEH may contribute to pulmonary hypertension. Anti-inflammatory, antiapoptotic, and proangiogenic actions of the EETs have also been reported.
Isoeicosanoids
The isoeicosanoids, a family of eicosanoid isomers, are formed nonenzymatically by direct free radical-based action on AA and related lipid substrates. The isoprostanes thus formed are prostaglandin stereoisomers. Because prostaglandins have many asymmetric centers, they have a large number of potential stereoisomers. COX is not needed for the formation of the isoprostanes, and its inhibition with aspirin or other NSAIDs should not affect the isoprostane pathway. The primary epimerization mechanism is peroxidation of arachidonate by free radicals. Peroxidation occurs while arachidonic acid is still esterified to the membrane phospholipids. Thus, unlike prostaglandins, these stereoisomers are “stored” as part of the membrane. They are then cleaved by phospholipases, circulate, and are excreted in urine. Isoprostanes are present in relatively large amounts (tenfold greater in blood and urine than the COX-derived prostaglandins). They have potent vasoconstrictor effects when infused into renal and other vascular beds and may activate prostanoid receptors. They also may modulate other aspects of vascular function, including leukocyte and platelet adhesive interactions and angiogenesis. It has been speculated that they may contribute to the pathophysiology of inflammatory responses in a manner insensitive to COX inhibitors. A particular difficulty in assessing the likely biologic functions of isoprostanes—several of which have been shown to serve as incidental ligands at prostaglandin receptors—is that while high concentrations of individual isoprostanes may be necessary to elicit a response, multiple compounds are formed coincidentally in vivo under conditions of oxidant stress. Analogous leukotriene and EET isomers have been described.
 BASIC PHARMACOLOGY OF EICOSANOIDS
BASIC PHARMACOLOGY OF EICOSANOIDS
MECHANISMS & EFFECTS OF EICOSANOIDS
Receptor Mechanisms
As a result of their short half-lives, the eicosanoids act mainly in an autocrine and a paracrine fashion, ie, close to the site of their synthesis, and not as circulating hormones. These ligands bind to receptors on the cell surface, and pharmacologic specificity is determined by receptor density and type on different cells (Figure 18–4). A single gene product has been identified for each of the PGI2 (IP), PGF2α (FP), and TXA2 (TP) receptors, while four distinct PGE2 receptors (EPs 1–4) and two PGD2 receptors (DP1 and DP2) have been cloned. Additional isoforms of the human TP (α and β), FP (A and B), and EP3 (I, II, III, IV, V, VI, e, and f) receptors can arise through differential mRNA splicing. Two receptors exist for LTB4 (BLT1 and BLT2) and for LTC4/LTD4 (cysLT1 and cysLT2). It appears that LTE4 functions through one or more receptors distinct from cysLT1/cysLT2, with some evidence that the orphan receptor GPR99 and the ADP receptor P2Y12 may function as LTE4 receptors. The formyl peptide (fMLP)-1 receptor can be activated by lipoxin A4 and consequently has been termed the ALX receptor. Receptor heterodimerization has been reported for a number of the eicosanoid receptors, providing for additional receptor subtypes from the currently identified gene products. All of these receptors are G protein-coupled; properties of the best-studied receptors are listed in Table 18–1.
TABLE 18–1 Eicosanoid receptors.1
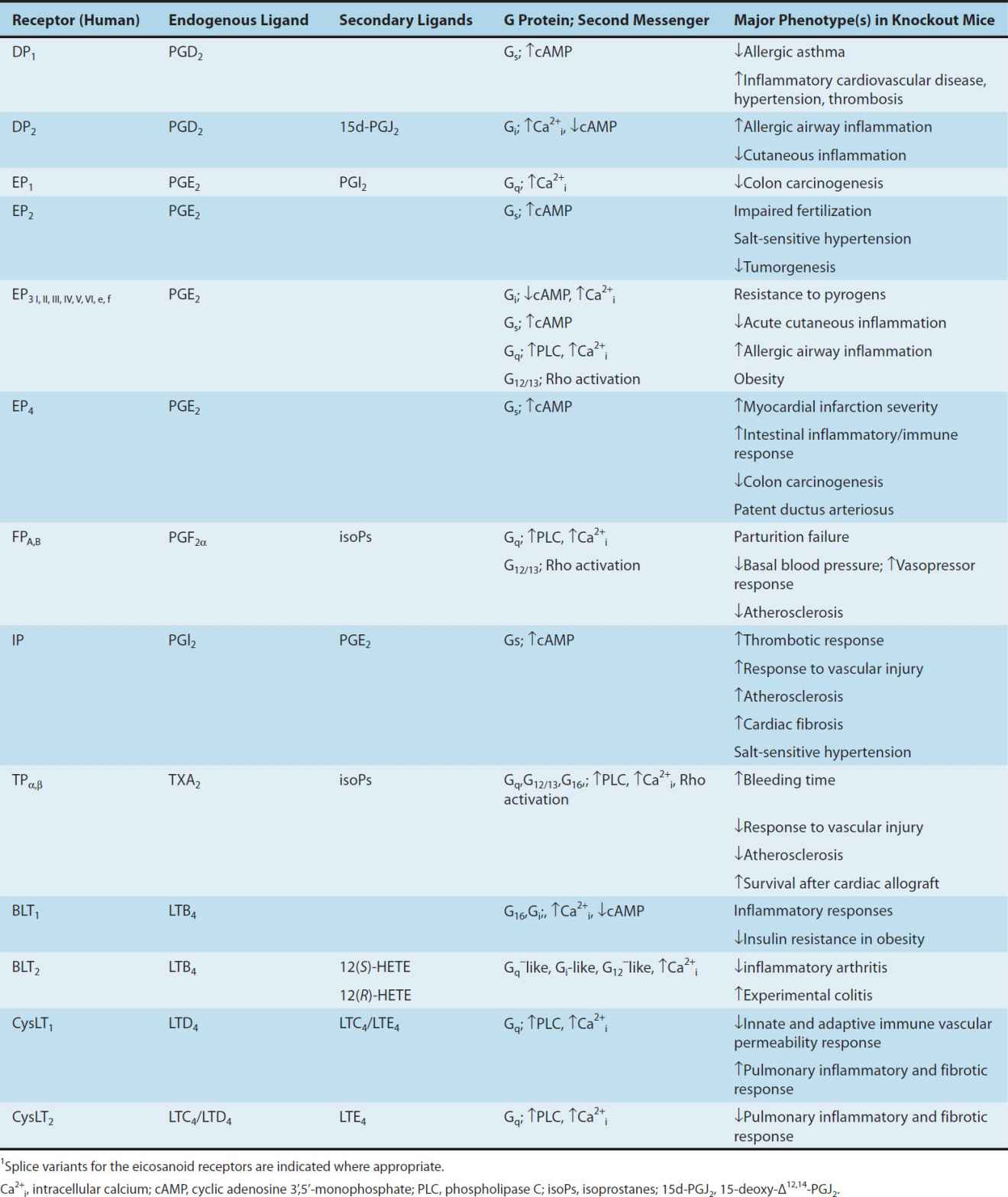
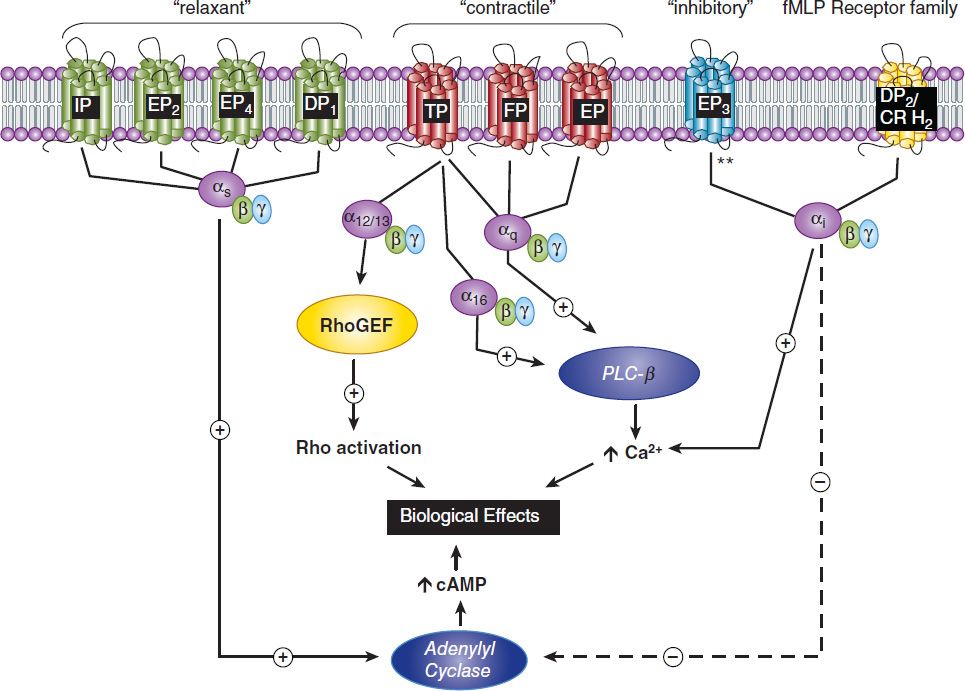
FIGURE 18–4 Prostanoid receptors and their signaling pathways. fMLP, formylated MetLeuPhe, a small peptide receptor; PLC-β, phospholipase C-β. All of the receptors shown are of the seven-transmembrane, G protein-coupled type. The terms “relaxant,” “contractile,” and “inhibitory” refer to the phylogenetic characterization of their primary effects. **, all EP3 isoforms couple through Gi but some can also activate Gs or G12/13 pathways. RhoGEF, rho guanine nucleotide exchange factor. See text for additional details.
EP2, EP4, IP, and DP1 receptors activate adenylyl cyclase via Gs. This leads to increased intracellular cAMP levels, which in turn activate specific protein kinases (see Chapter 2). EP1, FP, and TP activate phosphatidylinositol metabolism, leading to the formation of inositol trisphosphate, with subsequent mobilization of Ca2+ stores and an increase of free intracellular Ca2+. TP also couples to multiple G proteins, including G12/13 and G16, to stimulate small G protein signaling pathways, and may activate or inhibit adenylyl cyclase via Gs (TPα) or Gi (TPβ), respectively. EP3 isoforms can couple to both increased intracellular calcium and to increased or decreased cAMP. The DP2 receptor (also known as the chemoattractant receptor-homologous molecule expressed on TH2 cells, or CRTH2), which is unrelated to the other prostanoid receptors, is a member of the fMLP receptor superfamily. This receptor couples through a Gi-type G protein and leads to inhibition of cAMP synthesis and increases in intracellular Ca2+ in a variety of cell types.
LTB4 also causes inositol trisphosphate release via the BLT1 receptor, causing activation, degranulation, and superoxide anion generation in leukocytes. The BLT2 receptor, a low-affinity receptor for LTB4, is also bound with reasonable affinity by 12(S)- and 12(R)-HETE, although the biologic relevance of this observation is not clear. CysLT1 and cysLT2 couple to Gq, leading to increased intracellular Ca2+. Studies have also placed Gi downstream of cysLT2. An orphan receptor, GPR17, binds cysLTs and may negatively regulate the function of cysLT1, but its physiologic role remains ill defined. As noted above, the EETs promote vasodilation via paracrine activation of calcium-activated potassium channels on smooth muscle cells leading to hyperpolarization and relaxation. This occurs in a manner consistent with activation of a Gs-coupled receptor, although a specific EET receptor has yet to be identified. EETs may also act in an autocrine manner directly activating endothelial transient receptor potential channels to cause endothelial hyperpolarization, which is then transferred to the smooth muscle cells by gap junctions or potassium ions. Specific receptors for isoprostanes have not been identified, and the biologic importance of their capacity to act as incidental ligands at prostaglandin receptors remains to be established.
Although prostanoids can activate peroxisome proliferator-activated receptors (PPARs) if added in sufficient concentration in vitro, it remains questionable whether these compounds ever attain concentrations sufficient to function as endogenous nuclear-receptor ligands in vivo.
Effects of Prostaglandins & Thromboxanes
The prostaglandins and thromboxanes have major effects on smooth muscle in the vasculature, airways, and gastrointestinal and reproductive tracts. Contraction of smooth muscle is mediated by the release of calcium, while relaxing effects are mediated by the generation of cAMP. Many of the eicosanoids’ contractile effects on smooth muscle can be inhibited by lowering extracellular calcium or by using calcium channel-blocking drugs. Other important targets include platelets and monocytes, kidneys, the central nervous system, autonomic presynaptic nerve terminals, sensory nerve endings, endocrine organs, adipose tissue, and the eye (the effects on the eye may involve smooth muscle).
A. Smooth Muscle
1. Vascular—TXA2 is a potent vasoconstrictor. It is also a smooth muscle cell mitogen and is the only eicosanoid that has convincingly been shown to have this effect. The mitogenic effect is potentiated by exposure of smooth muscle cells to testosterone, which up-regulates smooth muscle cell TP expression. PGF2α is also a vasoconstrictor but is not a smooth muscle mitogen. Another vasoconstrictor is the isoprostane 8-iso-PGF2α, also known as iPF2αIII, which may act via the TP receptor.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


