successful completion of the following steps in cardiovascular development. Appropriate pathologic evaluation of CHD specimens will independently evaluate the situs and other structural components of the specimen (see Classification section below).
TABLE 13-1 CONGENITAL HEART DEFECTS: MAJOR CLINICAL FINDINGS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 13-2 SEQUENTIAL EXAMINATION OF HEART SEGMENTS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
great vessels the most common associated malformations. On the flip side, 11% of hearts with tricuspid atresia and 3% of hearts with D-transposition exhibit left-sided juxtaposition of the atrial appendages (18).
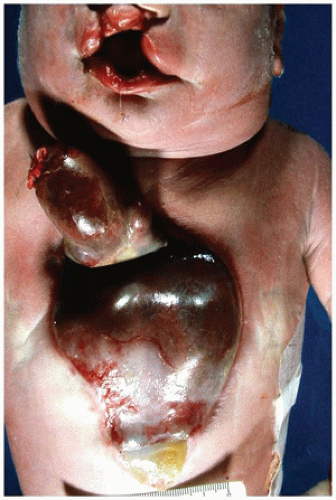 FIGURE 13-1 • Infant with multiple congenital anomalies including cleft lip seen at the top of the photograph and an anterior defect in the chest and abdomen through which the heart and liver protrude. |
In the more common anomaly, partial anomalous pulmonary venous connection, blood from one or more, but not all, of the pulmonary veins drains into a systemic vein or right atrium. This anomalous drainage is right sided in more than 80% of cases and most frequently enters the superior vena cava or the right atrium (14,22). More than 80% of cases occur in the setting of sinus venosus atrial septal defects (ASDs) as described earlier. The scimitar syndrome represents a variant of partial anomalous pulmonary venous connection characterized by anomalous pulmonary venous drainage into the inferior vena cava with a variety of associated cardiopulmonary anomalies. The most frequent associations include right lung hypoplasia, dextrocardia, systemic arterial supply to the lung, and abnormal bronchial anatomy (23).
TABLE 13-3 PULMONARY VENOUS MALFORMATIONS | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
TABLE 13-4 TOTAL ANOMALOUS PULMONARY VENOUS CONNECTION: CLASSIFICATION | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
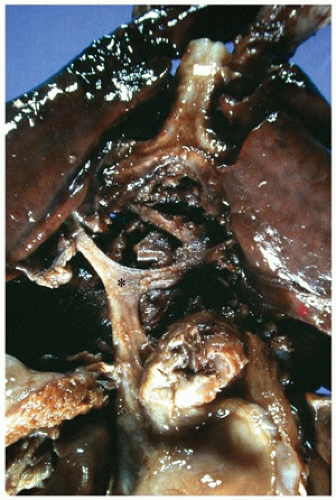 FIGURE 13-2 • Total anomalous pulmonary venous connection, infradiaphragmatic type, seen from the posterior view. A confluence of the pulmonary veins (asterisk) is isolated from the left atrium and drains into a vertical vein. This vein traverses the diaphragm to enter the portal venous system of the liver. |
a variably sized opening, which results in most instances in pulmonary venous obstruction. The foramen ovale may open into either compartment; when the opening is proximal to the obstruction, it can function as an escape valve for the pulmonary venous obstruction (14,27).
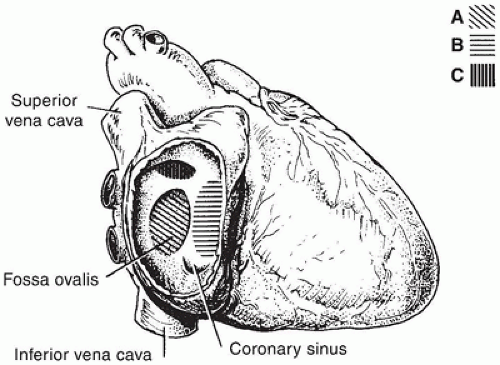 FIGURE 13-3 • The positions of various ASDs from the perspective of the right atrium, which has been opened laterally. A: Secundum defect. B: Primum defect. C: Sinus venosus defect. |
TABLE 13-5 TYPES OF ATRIAL SEPTAL MALFORMATION | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
ventricular outflow tract just beneath the aortic valve (Figure 13-5). In the right ventricle, they reside beneath the crista supraventricularis and behind the papillary muscle of the conus, partially obscured by the septal leaflet of the tricuspid valve (36) (Figure 13-6).
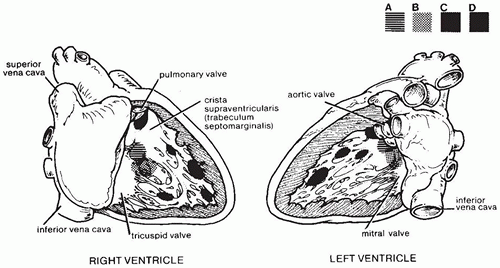 FIGURE 13-4 • The positions of the ventricular septal components and the corresponding VSTs from the lateral perspectives of the opened right and left ventricles. A: Membranous septum and perimembranous defect. B: Inlet septum and defect. C: Trabecular septum and trabecular muscular defects. D: Outlet septum and defect. |
TABLE 13-6 VENTRICULAR SEPTAL DEFECTS: CLASSIFICATION | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
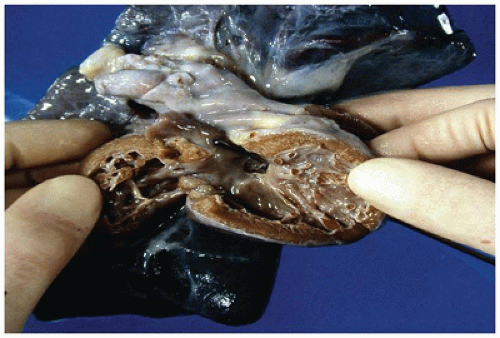 FIGURE 13-5 • Ventricular septal defect. An opened left ventricle with the free wall reflected laterally contains a perimembranous defect, visible in the outflow tract inferior to the aortic valve. The probe visible in the right ventricle in Figure 13-6 traverses the defect opening. |
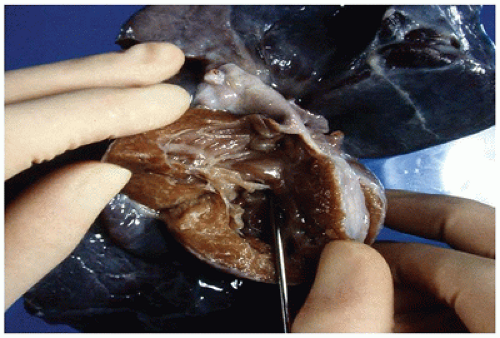 FIGURE 13-6 • Ventricular septal defect. An opened right ventricle with the free wall reflected superiorly contains a perimembranous defect hiding beneath the septal leaflet of the tricuspid valve. The probe traversing the defect is visible from the left ventricular aspect in Figure 13-5. |
TABLE 13-7 VENTRICULAR SEPTAL DEFECTS: CLINICAL GROUPS | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
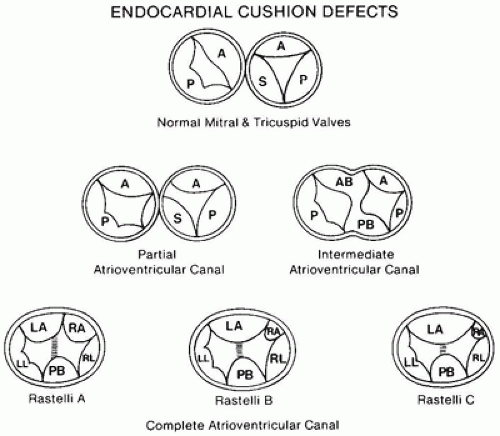 FIGURE 13-7 • Diagrammatic representation of the atrioventricular valves as viewed from the atria in a normal heart and with various atrioventricular septal defects. A, anterior leaflet; P, posterior leaflet; S, septal leaflet; AB, anterior bridging leaflet; PB, posterior bridging leaflet; LA, left anterior leaflet; RA, right anterior leaflet; RL, right lateral leaflet; LL, left lateral leaflet. |
A decrement of tissue at the crest of the ventricular inlet, lending the inlet septum a “scooped out” appearance
Elongation of the left ventricular outflow tract, which creates the “gooseneck” deformity
Abnormal formation of the AV valves, with a characteristic “cleft” in the left-sided anterior leaflet
valves insert, commissure-like, onto the rim of the ventricular septum; a connecting tongue of valve tissue covers the ventricular septum and closes the ring. The size of the defect in the ventricular septum also varies in these hearts, and in some cases, the valve leaflets may be less firmly adherent to the ventricular septum so that some interventricular shunting occurs (44).
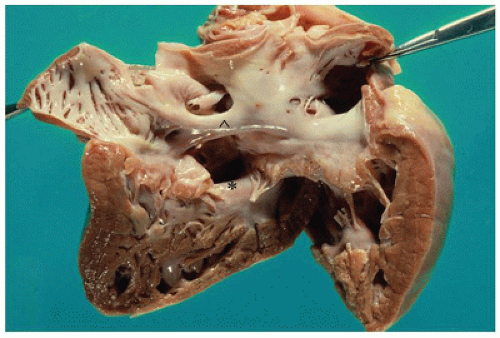 FIGURE 13-8 • Complete atrioventricular septal defect. A complete atrioventricular septal defect is readily visible centrally in this opened left atrium and ventricle. At the upper rim of the defect, a band of atrial septal tissue marked by the ˆ separates the upper secundum ASD from the lower ostium primum ASD. The lower rim of the defect marked by the * represents the upper rim of the ventricular septum. The anterior and the posterior bridging leaflets of the common AV valve extend over the defect without chordal insertion. |
TABLE 13-8 ATRIOVENTRICULAR SEPTAL DEFECTS: ASSOCIATED CARDIAC ANOMALIES AND SYNDROMES | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
TABLE 13-9 TYPES OF CONOTRUNCAL MALFORMATIONS | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
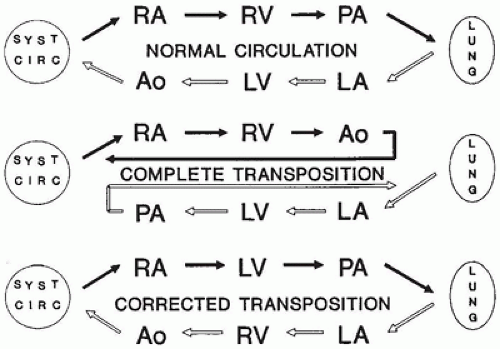 FIGURE 13-9 • Diagrammatic representation of normal blood flow (top), blood flow through complete transposition (middle), and blood flow through “corrected” transposition (bottom). Syst circ, systemic circulation; RA, right atrium; RV, right ventricle; PA, pulmonary artery; LA, left atrium; LV, left ventricle; Ao, aorta. |
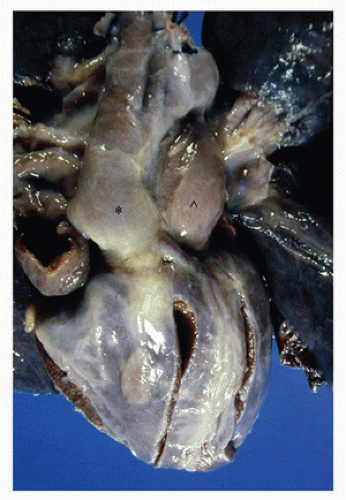 FIGURE 13-10 • Complete transposition of the great vessels from the anterior aspect of the heart. The aorta, marked with *, is situated to the right and slightly anterior to the pulmonary artery, marked with ˆ. The two vessels ascend in a parallel course. |
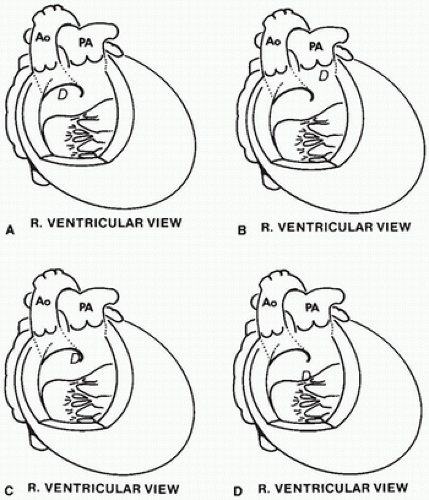 FIGURE 13-11 • The sites, D, of the VSDs in a DORV. A: Subaortic (60% to 65% of total cases). B: Subpulmonic (25% to 30%). C: Doubly committed (5% to 15%). D: Remote (10% to 15%). Ao, aorta; PA, pulmonary artery. |
TABLE 13-10 DOUBLE-OUTLET RIGHT VENTRICLE: RELATIONSHIP OF GREAT ARTERIES | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||
obstruction and DOLV with subpulmonic VSD by aortic outflow tract obstruction (63,64).
TABLE 13-11 DOUBLE-OUTLET RIGHT VENTRICLE: CLINICOPATHOLOGIC CATEGORIES | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
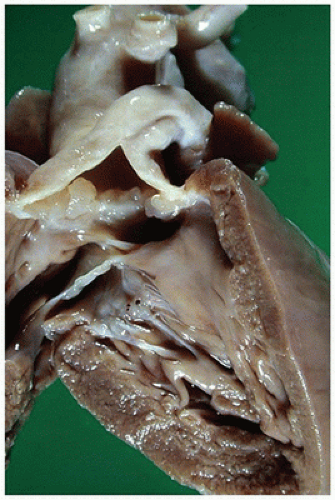 FIGURE 13-12 • Truncus arteriosus. The left ventricle free wall has been lifted to uncover the smooth-surfaced left ventricular outflow tract with a VSD opening at the top. Above the VSD lies a somewhat nodular truncus arteriosus valve. The main pulmonary artery almost immediately branches to the left from the common trunk; the aorta continues ascending posteriorly. |
the same general region as persistent truncus arteriosus, it is not seen in the chromosome 22q11 deletion syndromes (70).
TABLE 13-12 TRUNCUS ARTERIOSUS CLASSIFICATION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
other associated cardiovascular defects, most commonly pulmonary valvular stenosis, pulmonary atresia, or a VSD, occur in 30% to 40% of cases (73,75).
TABLE 13-13 TRICUSPID ATRESIA: CLINICAL CLASSIFICATION | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
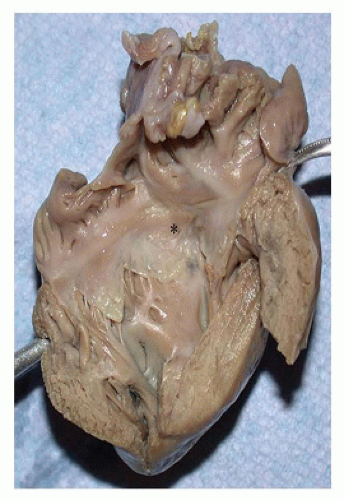 FIGURE 13-13 • Mild form of Ebstein anomaly. The opened right atrium and right ventricle display a markedly thickened ventricular wall. The septal (asterisk) and posterior leaflets of the tricuspid valve are fixed to the underlying ventricular wall. |
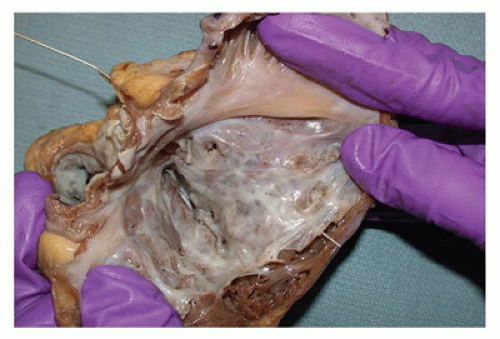 FIGURE 13-14 • Severe form of Ebstein anomaly. The opened right atrium uncovers a markedly enlarged and dysplastic anterior tricuspid leaflet attached to the apical myocardium by tiny chordae. A probe placed in the pulmonary artery traverses the remaining right ventricular cavity and appears at the base of this dysplastic valve, illustrating the severe obstruction to pulmonary inflow and outflow created by this defect. |
TABLE 13-14 MITRAL VALVE MALFORMATIONS | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
Associated with normally related great vessels and a shortened inlet septum (i.e., forme fruste of an AV septal defect)
Associated with TGA or DORV and a normal inlet septum
transposed in the vast majority of cases but may be normally related, atretic, or in a double-outlet configuration.
TABLE 13-15 UNIVENTRICULAR ATRIOVENTRICULAR CONNECTION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
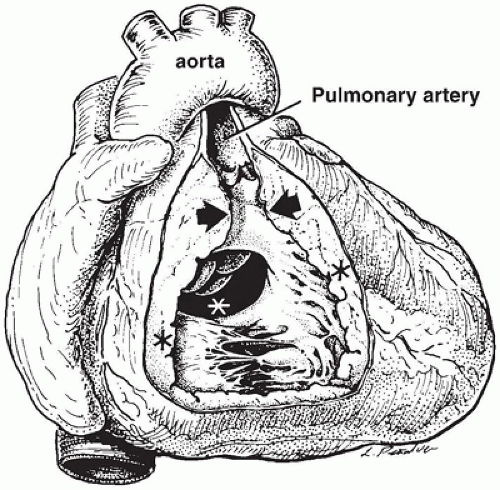 FIGURE 13-15 • An opened anterior right ventricle illustrates the four primary features of TOF: marked narrowing of the pulmonary infundibulum (between arrows); a large perimembranous VSD (white asterisk); dextroposed overriding aorta, visible through the VSD; and hypertrophy of the right ventricular myocardium (black asterisk). |
A complete AV septal defect accompanies TOF in 1% to 2% of cases, most often in children with trisomy 21.
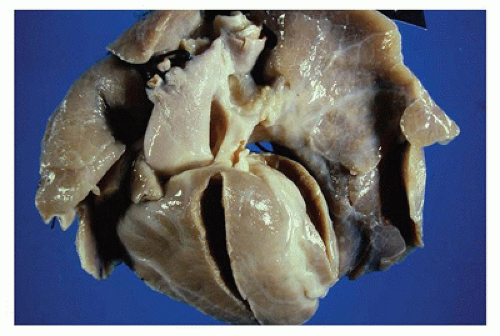 FIGURE 13-16 • Heart and lungs removed at autopsy with an unrepaired TOF. An incision through the anterior right ventricle ends at the base of a small pulmonary artery. The markedly enlarged aorta arises behind and to the right of the pulmonary artery. |
TABLE 13-16 PREVALENCE OF CONGENITAL HEART DEFECTS | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
for approximately one-third of cases (104) (Table 13-17). Associated cardiac malformations, present in approximately 66% of cases, include VSD, ASD, valvular pulmonary stenosis, and TOF (104,105). A variety of malformation syndromes include pulmonary artery stenosis (Table 13-18).
TABLE 13-17 SUPRAVALVULAR AND PERIPHERAL PULMONARY ARTERY STENOSIS | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
TABLE 13-18 PULMONARY ARTERY STENOSIS-ASSOCIATED MALFORMATION SYNDROMES | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


