INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should be able to:
Outline the changes in globin gene expression that occur during fetal development.
Explain the molecular genetics and inheritance of alpha and beta thalassemias.
Grasp the cellular pathogenesis of alpha and beta thalassemias.
Describe the clinical and laboratory features of beta thalassemia minor (trait) and beta thalassemia major and the different types of alpha thalassemia.
Understand the pathophysiologic principles underlying treatment of beta thalassemia major.
The thalassemias are an inherited group of disorders in which mutations in genes expressing alpha globin or beta globin result in impaired hemoglobin synthesis and microcytic anemia of varying severity. The thalassemias are subdivided into alpha (α) or beta (β) according to which globin genes are defective. Heterozygotes are generally asymptomatic, whereas individuals who inherit thalassemia genes from each parent often have life-threatening clinical manifestations. The thalassemias have attracted worldwide interest and attention because of their high prevalence and clinical importance. Moreover, an ever-expanding body of information on the molecular pathogenesis of the thalassemias has provided critical insights into fundamental problems in biology, particularly tissue-specific and development-specific gene regulation.
THE GLOBIN GENES
A diagram of the layout of the human globin genes is shown in Figure 8-1. A tandem pair of α-globin genes is located on chromosome 16, downstream from two embryonic α-like genes called zeta (ζ). The high homology of the α-globin genes leads to frequent unequal meiotic crossover events, which are the basis for the deletions that cause the α-thalassemias, discussed at the end of this chapter.
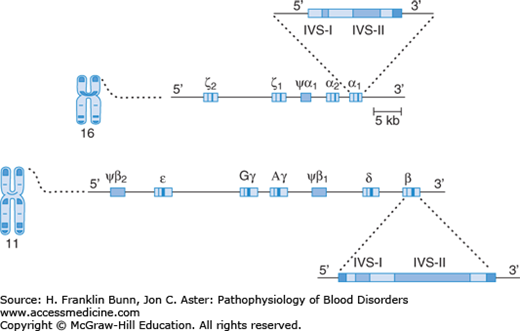
The β-globin gene is a member of a family located on chromosome 11. As in the α-globin gene family, epsilon (∊), the most 5′ (upstream) of these genes, is expressed only in early embryos. Downstream from this gene are two tandem gamma (γ) genes whose product is found in fetal hemoglobin (Hb F, α2β2), the hemoglobin that predominates throughout most of gestation. The delta (δ) gene product forms a minor hemoglobin component, Hb A2 (α2β;2), which has no functional importance but is useful in the diagnosis of the thalassemias (discussed later in this chapter.) The most 3′ (downstream) member of the family is the β gene whose product combines with α-globin to form Hb A (α2β2), the major hemoglobin component of adult red cells.
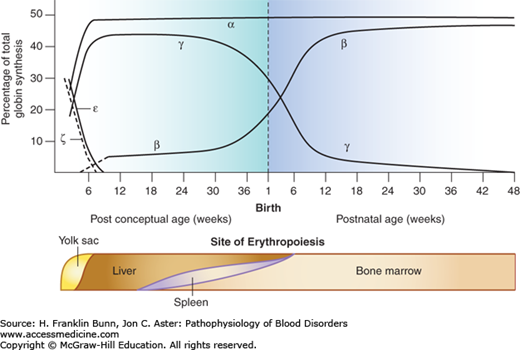
Figure 8-2 shows the expression of globin genes during development. Throughout gestation there is coordinate synthesis of globin products from the two chromosomes, permitting the sequential and orderly production of functional tetrameric hemoglobins. In the transition from embryo to fetus to birth, the globin genes on both chromosomes are “read” sequentially from left to right (5′ to 3′). During the first month of gestation, embryonic hemoglobins (ζ2∊2, ζ2γ2, ζ2γ2) are formed in erythroid cells located primarily in the yolk sac. During the remainder of fetal life, the sites of erythropoiesis gradually shift from the liver and spleen to the bone marrow. Fetal red cells contain predominantly Hb F (α2γ2). Shortly before birth, there is a dramatic switch from γ-globin to β-globin expression, which is complete by the time an infant is 6 to 8 months old. From then on, over 95% of the hemoglobin in normal red cells is adult Hb A (α2β2). The remaining hemoglobin consists of two minor components, Hb A2 and Hb F. The molecular basis of the switch from γ- to β-globin synthesis remains one of the foremost problems in hematology research and, as discussed later, impacts importantly the pathogenesis and treatment of β-thalassemia as well as of sickle cell disease (Chapter 9).
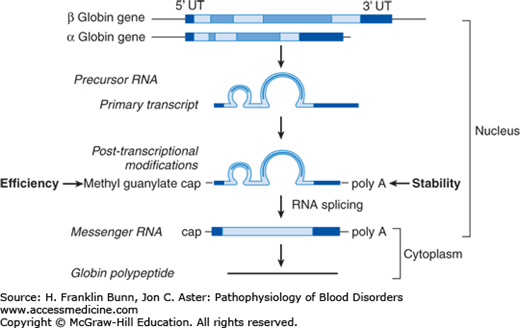
During their maturation, erythroid cells become increasingly dedicated to the production of hemoglobin. Far upstream of the globin genes are regulatory elements called locus control regions at which erythroid-specific transcription factors bind cooperatively to response elements in the DNA to assure tissue-specific, high-level, and closely matched transcription of α and β (or γ) globin ribonucleic acids (RNAs). Like other precursor RNAs, globin RNA must be processed into messenger RNA (mRNA) and transported from the nucleus to cytoplasm for translation (protein synthesis) to occur. All globin genes have three exons and two introns. As shown in Figure 8-3, precursor RNA processing involves sequential splicing of the two introns followed by capping at the 5′ end of the RNA, which greatly enhances translation efficiency, along with cleavage and polyadenylation of the 3′ untranslated region, which enhances mRNA stability. Mutations at critical sites involved in splicing, capping, and polyadenylation can result in defective globin synthesis and therefore thalassemia.
FIGURE 8-3
Processing of ribonucleic acid (RNA) expressed by the a-globin gene. UT, untranslated. (Modified with permission from Chapter 21. MJ Cunningham, VG Sankaran, DG Nathan, SH Orkin. The Thalassemias, in: Orkin SH, Nathan DG, eds. Nathan and Oski’s Hematology of Infancy and Childhood. 5th ed. Philadelphia, PA, Saunders; 2008: 1019.)
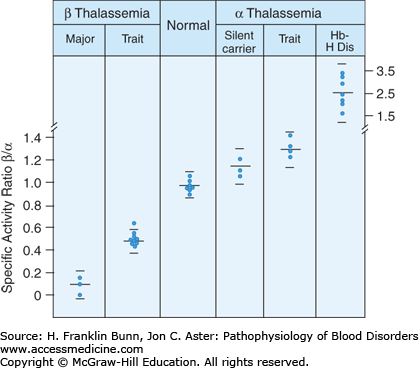
As erythroid precursors reach full maturation, over 95% of protein synthesis is dedicated to globin production. This remarkable up-regulation is matched by markedly enhanced synthesis of heme. In erythroblasts and reticulocytes, globin released from the polyribosome combines with heme to form subunits capable of assembly into heterotetramers such as Hb A (α2β2). This assembly depends on balanced synthesis of α and β subunits. As shown in Figure 8-4, globin chain synthesis is imbalanced in the thalassemias. The presence of excess free globin chains is an important contributor to the pathogenesis of these disorders.
FIGURE 8-4
The β/α globin biosynthetic ratio in different forms of thalassemia. Peripheral blood was incubated with a radiolabeled amino acid, followed by separation of globin subunits. (Modified with permission from Nathan DG. Thalassemia. N Engl J Med. 1972;286:586. Copyright © 1972 Massachusetts Medical Society, all rights reserved.)
β-THALASSEMIA
As shown in Figure 8-1, chromosome 11 contains a single β-globin gene. Individuals who inherit a β-thalassemia mutation from one parent and a normal β-globin gene from the other are heterozygotes, often designated as having β-thalassemia trait or β-thalassemia minor. Individuals who inherit the same β-thalassemia mutation from each parent are true homozygotes. Because multiple types of mutations can give rise to β-thalassemia, an individual often inherits a different mutation from each parent and is therefore a compound heterozygote. If homozygotes or compound heterozygotes have severe disease, they are designated as having β-thalassemia major or Cooley anemia, whereas if they have milder clinical manifestations, they have β-thalassemia intermedia.
Over 100 million people in the world are heterozygous carriers of a β-thalassemia mutation, nearly two-thirds from Asia and the rest divided between Africa, Europe, and the Americas. Affected individuals come primarily from tropical areas. Reasonably convincing evidence suggests that β-thalassemia heterozygotes are protected against infantile falciparum malaria, which is often fatal. Thus natural selection has enabled β-thalassemia genes to gradually rise to high levels in populations where malaria is endemic.
The β-thalassemias are caused by diverse mutations involving the promoter, coding sequences, intron-exon boundaries, and the polyadenylation site of the β-globin gene. The mutant alleles are conveniently subdivided into two groups: β0, in which no β-globin is detectable, and β+, in which a small amount of normal β-globin protein is produced. Most β0-thalassemia alleles involve single base substitutions in the coding region of the β polypeptide that introduce premature stop codons or small insertions or deletions that cause shifts in the reading frame of the mRNA. In either case, the resulting truncated polypeptide is dysfunctional and so unstable that it is undetectable in the patient’s red cells. Other less common genetic mechanisms that result in β0 alleles include deletions and mutations at splice junctions.
In β+-thalassemia, the defective gene permits the production of normal β-globin, but the amount is markedly reduced. This type of β-thalassemia is usually due to a single base substitution that either creates a new (false) splice site or lowers the efficiency of a normal splice site. In both instances, some normal splicing occurs, and some normal β-globin is made, albeit in decreased amounts. When the alternate splice site is used, a nonsense mRNA is produced that cannot form a stable or useful protein product. Less often β+-thalassemia is caused by mutations in the 5′ promoter or in a 3′ site in β-globin RNA where it is cleaved prior to polyadenylation.



