Targeted Therapies: Tyrosine Kinase Inhibitors, Monoclonal Antibodies, and Cytokines
Many new agents are available to block the fundamental mutations that cause specific cancers: aberrant growth factor receptors, dysregulated intracellular signaling pathways, defective DNA repair and apoptosis, and tumor angiogenesis. The primary tools for inhibiting these targets are monoclonal antibodies that attack cell surface receptors and antigens, and synthetic small molecules that enter cells and engage critical enzymes. These 2 classes of drugs have very different pharmacological properties.
Monoclonal antibodies kill tumor cells by blocking cell surface receptor function and by recruiting immune cells and complement to the antigen–antibody complex. They may be armed to carry toxins or radionuclides to the cells of interest, thereby enhancing their cytotoxic effects. They generally are specific for a single receptor, have a long plasma t1/2, and require only intermittent administration. Small molecules may attack the same targets and pathways as the monoclonals, but may also exert their effect by entering cells and inhibiting enzymatic functions (usually tyrosine kinase reactions). The small molecules often inhibit multiple enzymatic sites, have a broad spectrum of target kinases, and tend to be substrates of hepatic CYPs with a t1/2 of 12-24 h, and thus require daily oral administration.
These 2 drug classes, when targeted against the same pathway, may have significantly different spectra of antitumor activity. Thus, monoclonal antibodies to the epidermal growth factor receptor (EGFR) are effective in the treatment of head and neck and colon cancers, while small molecules, such as erlotinib and gefitinib, attack the intracellular tyrosine kinase function of the same receptor and have a different spectrum of antitumor activity (non–small cell lung cancer). The specific drug target is of central importance in cancer chemotherapy and forms the organizational basis for the discussion below.
PROTEIN TYROSINE KINASE INHIBITORS
There are 3 basic types of protein kinases (see Chapter 3):
• Kinases that specifically phosphorylate tyrosine residues
• Kinases that phosphorylate serine and threonine residues
• Kinases with activity toward all 3 residues
Tyrosine kinases can be further subdivided into those with an extracellular ligand-binding domain (receptor tyrosine kinases, associated with growth factor receptors, Figure 62–1) and intracellular enzymes (nonreceptor tyrosine kinases, e.g., src, abl, jak, fak, srm). In a number of human malignancies, mutations that constitutively activate protein tyrosine kinases are implicated in malignant transformation.
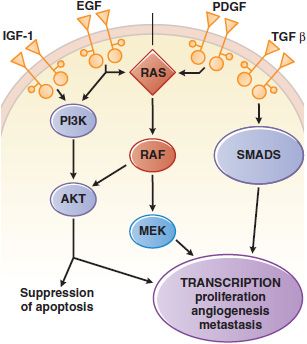
Figure 62–1 Growth factor signaling. Binding of agonist ligands to growth factor receptors (monospanning membrane proteins) causes receptor dimerization and activation of cytosolic protein kinase domains, leading to activation of multiple signaling pathways. Shown here are the RAS/MAPK/ERK, PI3K, and SMAD pathways, each of which is activated by receptors or cross-talk from adjacent pathways. Their signals regulate proliferation, metabolism, survival, and the synthesis of other growth factors, such as the vascular endothelial growth factor (VEGF).
INHIBITORS OF THE BCR-ABL KINASE: IMATINIB, DASATINIB, AND NILOTINIB
Imatinib mesylate (STI 571, GLEEVEC, GLIVEC) targets the BCR-ABL tyrosine kinase, which underlies chronic myelogenous leukemia (CML). A single molecular event, in this case the 9:22 translocation, leads to expression of the Abelson proto-oncogene kinase ABL fused to BCR (breakpoint cluster region), yielding a constitutively activated protein kinase, BCR-ABL, and then the malignant phenotype.
Imatinib and the related compounds dasatinib and nilotinib induce clinical and molecular remissions in >90% of CML patients in the chronic phase of disease. Imatinib treats other tumors that carry related tyrosine kinase mutations, including GI stromal tumors (driven by c-KIT mutation), and hypereosinophilia syndrome, chronic myelomonocytic leukemia, and dermatofibrosarcoma protuberans (all driven by mutations that activate the PDGF receptor [PDGFR]).
CHARACTERISTICS AND MECHANISM OF ACTION. Imatinib was identified through high-throughput screening against the BCR-ABL kinase. Dasatinib (BMS-354825, SPRYCEL), a second-generation BCR-ABL inhibitor, inhibits the Src kinase, and unlike imatinib, it binds both the open and closed configurations of the BCR-ABL kinase. Nilotinib (AMN107, TASIGNA) was designed to have increased potency and specificity compared to imatinib. Its structure overcomes mutations that cause imatinib resistance. Imatinib and nilotinib bind to a segment of the kinase domain that fixes the enzyme in a closed or nonfunctional state, in which the protein is unable to bind its substrate/phosphate donor, ATP. The 3 BCR-ABL kinase inhibitors differ in their potency of inhibition, their binding specificities, and their susceptibility to resistance mutations in the target enzyme. Dasatinib [(IC50)=<1 nM] and nilotinib [(IC50)=<20 nM] inhibit BCR-ABL kinase more potently than does imatinib [(IC50)=100 nM].
MECHANISMS OF RESISTANCE. Resistance to the tyrosine kinase inhibitors arises from point mutations in 3 separate segments of the kinase domain (see Figure 62–1 in the 12th edition of the parent text). The contact points between imatinib and the enzyme become sites of mutations in drug-resistant leukemic cells; these mutations prevent tight binding of the drug and lock the enzyme in its open configuration, in which it has access to substrate and is enzymatically active. Nilotinib retains inhibitory activity in the presence of most point mutations that confer resistance to imatinib. Other mutations affect the phosphate-binding region and the “activation loop” of the domain with varying degrees of associated resistance. Some mutations, such as at amino acids 351 and 355, confer low levels of resistance to imatinib, possibly explaining the clinical response of some resistant patients to dose escalation of imatinib.
Molecular studies have detected resistance-mediating kinase mutations prior to initiation of therapy, particularly in patients with Ph+ acute lymphoblastic leukemia (ALL) or CML in blastic crisis. This finding supports the hypothesis that drug-resistant cells arise through spontaneous mutation and expand under the selective pressure of drug exposure. Mechanisms other than BCR-ABL kinase mutation play a minor role in resistance to imatinib. Amplification of the wild-type kinase gene, leading to overexpression of the enzyme, has been identified in tumor samples from patients resistant to treatment. The multidrug resistant (MDR) gene confers resistance experimentally but has not been implicated in clinical resistance. Philadelphia chromosome-negative clones lacking the BCR-ABL translocation and displaying the karyotype of myelodysplastic cells may emerge in patients receiving imatinib for CML and may progress to myelodysplasia (MDS) and to acute myelocytic leukemia (AML). Their origin is unclear.
ADME
Imatinib. Imatinib is well absorbed after oral administration and reaches maximal plasma concentrations within 2-4 h. The elimination t1/2 of imatinib and its major active metabolite, the N-desmethyl derivative, are ~18 and 40 h, respectively. Food does not change the pharmacokinetic profile. Doses >300 mg/day achieve trough levels of 1 <M, which correspond to in vitro levels required to kill BCR-ABL–expressing cells. In the treatment of GI stromal cell tumors (GISTs), higher doses (600 mg/day) may improve response rates. CYP3A4 is the major metabolizer of imatinib; thus, drugs that induce or interact with CYP3A4 can alter the pharmacokinetics of imatinib. Coadministration of imatinib and rifampin, an inducer of CYP3A4, lowers the plasma imatinib AUC by 70%. Imatinib, as a competitive CYP3A4 substrate, inhibits the metabolism of simvastatin and increases its plasma AUC by 3.5-fold.
Dasatinib. Oral dasatinib is well absorbed; its bioavailability is significantly reduced at neutral gastric pH (i.e., after antacids and H2 blockers) but is unaffected by food. The plasma t1/2 of dasatinib is 3-5 h. Dasatinib exhibits dose proportional increases in AUC, and its clearance is constant over the dose range of 15-240 mg/day. Dasatinib doses of 70 mg twice a day, 100 mg daily, and 140 mg daily are equally effective in patients with CML, although the 100-mg daily dose improves progression-free survival. Dasatinib is metabolized primarily by CYP3A4, with minor contributions by FMO3 and UGT. Plasma concentrations of dasatinib are affected by inducers and inhibitors of CYP3A4 in a similar fashion to imatinib.
Nilotinib. Approximately 30% of an oral dose of nilotinib (400 mg 2 times daily) is absorbed after administration, with peak concentrations in plasma 3 h after dosing. Unlike the other BCR-ABL inhibitors, nilotinib’s bioavailability increases significantly in the presence of food. The drug has a plasma t1/2 17 h, and plasma concentrations reach a steady state only after 8 days of daily dosing. Nilotinib is metabolized by CYP3A4, with predictable alteration by inducers, inhibitors, and competitors of CYP3A4. Nilotinib is a substrate and inhibitor of P-glycoprotein.
THERAPEUTIC USES. These protein tyrosine kinase inhibitors have efficacy in diseases in which the ABL, kit, or PDGFR have dominant roles in driving the proliferation of the tumor, reflecting the presence of a mutation that results in constitutive activation of the kinase. Imatinib shows remarkable therapeutic benefits in patients with chronic-phase CML (BCR-ABL), GIST (kit mutation positive), chronic myelomonocytic leukemia (EVT6-PDGFR translocation), hypereosinophilia syndrome (FIP1L1-PDGFR), and dermatofibrosarcoma protuberans (constitutive production of the ligand for PDGFR). It is the agent of choice for GIST patients with metastatic disease and as adjuvant therapy of c-kit–positive GIST. The currently recommended dose of imatinib is 400-600 mg/day. Dasatinib is approved for patients with CML resistant or intolerant to imatinib in both chronic (100 mg/day) and advanced phases of disease (70 mg twice daily), and for use combined with cytotoxic chemotherapy in patients with Ph+ ALL who are resistant or intolerant to prior therapies. Nilotinib is approved for patients with CML resistant to or intolerant of prior imatinib therapy.
TOXICITY. Imatinib, dasatinib, and nilotinib cause GI symptoms (diarrhea, nausea, and vomiting) that are usually readily controlled. All 3 drugs promote fluid retention, edema, and peri-orbital swelling. Dasatinib may cause pleural effusions. Nilotinib may prolong the QT interval. Myelosuppression occurs infrequently but may require transfusion support, dose reduction, or discontinuation of the drug. These drugs can be associated with hepatotoxicity. Most nonhematological adverse reactions are self-limited and respond to dose adjustments. After the adverse reactions have resolved, the drug may be reinitiated and titrated back to effective doses.
EPIDERMAL GROWTH FACTOR RECEPTOR INHIBITORS
The EGFR belongs to the ErbB family of transmembrane receptor tyrosine kinases. EGFR, also known as ErbB1 or HER1, is essential for the growth and differentiation of epithelial cells. Ligand binding to the extracellular domain of EGFR family members causes receptor dimerization and stimulates the protein tyrosine kinase activity of the intracellular domain, resulting in autophosphorylation of several Tyr residues in the C-terminal domain. Recognition of the phosphotyrosines by other proteins initiates protein-protein interactions that result in stimulation of signaling pathways including MAPK, PI3K/Akt, and STAT pathways (see Figure 62–1). In epithelial cancers, overexpression and mutational activation of the EGFR are a common finding and create a dependence on EGFR signaling in these tumors.
Two separate classes of drugs that target the EGFR pathway are important agents in the therapy of solid tumors. The EGFR tyrosine kinase inhibitors erlotinib and gefitinib bind to the kinase domain and block the enzymatic function of EGFR. The monoclonal antibodies cetuximab and panitumumab bind specifically to the extracellular domain of EGFR and inhibit EGFR-dependent signaling.
GEFITINIB
Mechanism of Action. Gefitinib inhibits the EGFR tyrosine kinase by competitive blockade of ATP binding. Gefitinib has an IC50 of 20-80 nM for the EGFR tyrosine kinase but is significantly less potent against HER2 (ErbB2/neu). Gefitinib has antitumor activity in human xenograft tumors that exhibit high levels of EGFR expression.
ADME. Oral bioavailability is ~60%; peak plasma concentrations are achieved within 3-7 h. Absorption of gefitinib is not significantly altered by food but is reduced by drugs that cause elevations in gastric pH. Metabolism of gefitinib is predominantly via CYP3A4, with a terminal t1/2 of 41 h. Inducers of CYP3A4 activity decrease gefitinib plasma concentrations and efficacy; conversely, CYP3A4 inhibitors increase plasma concentrations.
Therapeutic Uses. Gefitinib initially was approved for the third-line treatment of patients with non–small cell lung cancer. However, a large clinical trial failed to show an effect on survival, leading the FDA to restrict its use to patients who have previously received clinical benefit from the drug. Two large trials have failed to demonstrate a benefit of gefitinib in combination with chemotherapy: patients who were nonsmokers, Asians, or women were most likely to respond to gefitinib. Tumors from these patients frequently have characteristic activating mutations in EGFR. The standard dose is 250 mg/day.
Adverse Effects and Drug Interactions. Diarrhea and pustular/papular rash occur in ~50% of patients. Other side effects include dry skin, nausea, vomiting, pruritus, anorexia, and fatigue. Most adverse effects occur within the first month of therapy and are manageable with medications and dose reductions. Asymptomatic increases in liver transaminases may necessitate dose reduction or discontinuation of therapy. Interstitial lung disease occurs in <2% of patients and may have a fatal outcome. Inducers and inhibitors of CYP3A4 will alter plasma concentrations. Patients using warfarin should be monitored closely for elevation of the international normalized ratio (INR) while taking gefitinib.
ERLOTINIB
Mechanism of Action. Erlotinib (TARCEVA) is a potent inhibitor of the EGFR tyrosine kinase, competitively inhibiting ATP binding at the active site of the kinase. Erlotinib has an IC50 of 2 nM for the EGFR kinase. Tumors harboring k-ras mutations and EML4-ALK translocations do not respond to EGFR kinase inhibitors.
ADME. Erlotinib is ~60% absorbed after oral administration but should not be taken with food, which increases its bioavailability to ~100%. Peak plasma levels occur after 4 h. Erlotinib has a t1/2 of 36 h and is metabolized by CYP3A4 and to a lesser extent by CYPs 1A2 and 1A1. The standard daily dose of erlotinib results in a plasma AUC ~10-fold greater than the AUC of gefitinib.
Therapeutic Uses. Erlotinib is approved for second-line treatment of patients with locally advanced or metastatic non–small cell lung cancer. Erlotinib also is approved for first-line treatment of patients with locally advanced, unresectable, or metastatic pancreatic cancer in combination with gemcitabine. The recommended dose of erlotinib in non–small cell lung cancer is a 150-mg tablet daily. In pancreatic cancer, the dose is a 100-mg tablet daily, taken at least 1 h before or 2 h after a meal.
Adverse Effects and Drug Interactions. The most common adverse reactions are diarrhea, an acneform rash, anorexia, and fatigue. Serious or fatal interstitial lung disease occurs with a frequency of 0.7-2.5%. Serious or fatal hepatic failure due to erlotinib has been reported, particularly in patients with baseline hepatic dysfunction. Other rare but serious toxicities include GI perforation, renal failure, arterial thrombosis, microangiopathic hemolytic anemia, hand-foot skin reaction, and corneal perforation or ulceration. Erlotinib therapy may cause rare cases of Stevens-Johnson syndrome/toxic epidermal necrolysis.
Concurrent use of proton-pump inhibitors decreases the bioavailability of erlotinib by 50%. Plasma levels can vary due to drug interactions with inducers or inhibitors of CYP3A4. Patients using warfarin may experience elevations of the INR while taking erlotinib. Smoking accelerates metabolic clearance of erlotinib and may decrease its antitumor effects.
RESISTANCE TO GEFITINIB AND ERLOTINIB
Patients with non–small cell lung cancer who initially respond to erlotinib or gefitinib have tumors that are dependent on the EGFR signaling pathway. Tumors containing mutations in EGFR initially respond to erlotinib and gefitinib but eventually progress. A secondary mutation in the EGFR gatekeeper residue, T790M, prevents binding of drug to the kinase domain and confers resistance. Other potential mechanisms of resistance include activation of downstream mediators, efflux of drug, and altered receptor trafficking. Therapy directed at the EGFR may delay disease progression in patients with non–small cell lung cancers that lack activating EGFR mutations, although response rates approach zero in these patients.
CETUXIMAB
Cetuximab (ERBITUX) is a recombinant chimeric human/mouse immunoglobulin G1 (IgG1) antibody that binds to the extracellular domain of EGFR. Such antibodies, although sharing the same target with erlotinib and gefitinib and having a similar side effect profile, have a different spectrum of antitumor activity.
Mechanism of Action. Cetuximab binds specifically to the extracellular domain of EGFR and prevents ligand-dependent signaling and receptor dimerization, thereby blocking cell growth and survival signals. Cetuximab also may mediate antibody-dependent cellular cytotoxicity against tumor cells.
ADME. Cetuximab exhibits nonlinear pharmacokinetics. Following intravenous administration, steady-state levels are achieved by the third weekly infusion. Therapeutic doses that saturate total body receptor pools of EGFR follow zero-order kinetics for elimination. Clearance occurs via EGFR binding and internalization and by degradation in the reticuloendothelial system.
Therapeutic Uses
Head and Neck Cancer. Cetuximab is used in combination with radiation therapy for locally or regionally advanced squamous cell carcinoma of the head and neck (HNSCC). It also is indicated in monotherapy for patients with metastatic or recurrent HNSCC who fail platinum-based chemotherapy. It is a useful agent in combination with cisplatin-based chemotherapy.
Metastatic Colon Cancer. Cetuximab is approved as a single agent for the treatment of EGFR-positive metastatic colorectal cancer; cetuximab is used in patients who cannot tolerate irinotecan-based therapy and in combination with irinotecan for patients refractory to oxaliplatin, irinotecan, and 5-fluorouracil (5-FU). In the first-line setting, cetuximab may improve survival in combination with 5-FU/leucovorin and irinotecan or oxaliplatin. About 40-50% of colorectal tumors carry mutations in the k-ras oncogene and are resistant to the effects of cetuximab. Cetuximab enhances the effectiveness of chemotherapy in patients with k-ras mutant tumors but not k-ras wild-type tumors. The standard dose of cetuximab is a single loading dose of 400 mg/m2 intravenously, followed by weekly doses of 250 mg/m2 intravenously for the duration of treatment.
Adverse Effects. Side effects include an acneform rash (in the majority of patients), pruritus, nail changes, headache, and diarrhea. Rare but serious adverse effects include cardiopulmonary arrest, interstitial lung disease, and hypomagnesemia. In addition, patients can develop anaphylactoid reactions during infusion, which may be related to preexisting IgE antibodies that are more prevalent in patients from the southern U.S.
PANITUMUMAB
Panitumumab (VECTIBIX) is a recombinant, fully humanized IgG2k antibody that binds specifically to the extracellular domain of EGFR. Unlike cetuximab, it does not mediate antibody-dependent cell-mediated cytotoxicity.
Panitumumab exhibits nonlinear pharmacokinetic characteristics. Following intravenous administration every 2 weeks, steady-state levels are achieved by the third infusion. The mean t1/2 is 7.5 days. Panitumumab improves progression-free survival in patients with metastatic colorectal carcinoma. The dose of panitumumab is 6 mg/kg intravenously given once every 2 weeks. The adverse effects with panitumumab are similar to cetuximab and include rash and dermatological toxicity, severe infusion reactions, pulmonary fibrosis, and electrolyte abnormalities.
HER2/NEU INHIBITORS
Both antibodies (trastuzumab, pertuzumab) and small molecules (lapatinib et al.) have striking antitumor effects in patients with HER2-positive breast cancer, and have become essential therapeutic agents in combination with cytotoxic chemotherapy for this aggressive malignancy.
TRASTUZUMAB. Trastuzumab (HERCEPTIN) is a humanized monoclonal antibody that binds to the external domain of HER2/neu (ErbB2).
Thirty percent of breast cancers overexpress this receptor due to gene amplification on chromosome 17. Amplification of the receptor is associated with lower response rates to hormonal therapies and to most cytotoxic drugs, with the exception of anthracyclines. Patients with HER2/neu-amplified tumors have higher recurrence rates after standard adjuvant therapy and poorer overall survival. The internal domain of the HER2/neu glycoprotein encodes a tyrosine kinase that activates downstream signal, enhances metastatic potential, and inhibits apoptosis. Trastuzumab exerts its antitumor effects through: inhibition of homo- or heterodimerization of receptor, thereby preventing receptor kinase activation and downstream signaling; initiation of Fcγ-receptor-mediated antibody-dependent cellular cytotoxicity; and blockade of the angiogenetic effects of HER2 signaling.
Therapeutic Uses. Trastuzumab is approved for HER2/neu-overexpressing metastatic breast cancer, in combination with paclitaxel as initial treatment or as monotherapy following chemotherapy relapse. Trastuzumab synergizes with other cytotoxic agents in HER2/neu-overexpressing cancers.
Pharmacokinetics and Toxicity. Trastuzumab has dose-dependent pharmacokinetics with a mean t1/2 of 5.8 days on the 2-mg/kg maintenance dose. Steady-state levels are achieved between 16 and 32 weeks. The infusional effects of trastuzumab are typical for monoclonal antibodies and include fever, chills, nausea, dyspnea, and rashes. Premedication with diphenhydramine and acetaminophen is indicated. The most serious toxicity of trastuzumab is cardiac failure; reasons for cardiotoxicity are poorly understood. Before initializing therapy, baseline electrocardiogram and cardiac ejection fraction measurement should be obtained to rule out underlying heart disease, and patients deserve careful clinical follow-up thereafter for signs or symptoms of congestive heart failure. When trastuzumab is used as a single agent, <5% of patients will experience a decrease in left-ventricular ejection fraction, and 1% will have clinical signs of congestive failure. Left-ventricular dysfunction occurs in up to 20% of patients who receive the antibody in combination with doxorubicin and cyclophosphamide. The risk of cardiac toxicity is greatly reduced with taxane–trastuzumab combinations.
LAPATINIB.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


