http://evolve.elsevier.com/McCuistion/pharmacology/
Genes, inflammatory processes, infection, environment, and diet can influence cancer cells. Cancer cells grow out of control and can spread to distant parts of the body, invading normal tissues to the extent that tissues and vital organs can no longer function normally.
As discussed in Chapter 32, traditional chemotherapy is systemic, and its chemical makeup is cytotoxic such that it directly damages or kills normal cells and cancer cells. Chemotherapeutic drugs inhibit mitosis, which can damage the RNA and DNA and thus prevent cell division, eventually causing apoptosis, or programmed cell death. Even though traditional chemotherapy treatments improve cancer control and increase long-term survival, adverse effects of traditional chemotherapy can be life-threatening. Furthermore, traditional chemotherapy’s cancer cell–killing effect is limited by the dosages and scheduling regimens needed to reduce toxic side effects on normal cells. Drugs that molecularly target cells associated with cancer are currently the focus of many research.
In 1998, targeted therapy was first recognized as a potential cancer therapy against certain cell receptors. Targeted therapy for cancer treatment differs from traditional cancer chemotherapy in that targeted therapies are specific, deliberate, and cytostatic, whereas most standard chemotherapies are not specific but rather are cytotoxic to normal and abnormal cells. Targeted therapy is the cornerstone of precision medicine because it directs the treatment according to the person’s genes and proteins. The National Cancer Institute (NCI) defines targeted therapy as drugs or other substances that block the growth and spread of cancer by interfering with specific molecules involved in tumor growth, progression, and spread.
The ability to treat cancers with targeted therapy continues to expand. Currently, there are three approaches to targeted therapy. One approach is comparing individual proteins in cancer cells with those of normal cells. An example of a differentially expressed target is the human epidermal growth factor receptor 2 protein (ERBB2), which is expressed at high levels in breast and gastric cancers. Targeted therapy directed at ERBB2 includes trastuzumab, which has been approved by the U.S. Food and Drug Administration (FDA) to treat certain breast and gastric cancers. A second approach is to identify mutant proteins that can cause cancer progression. An example of a mutant cell is the BRAF gene in its altered form, BRAF V600E, in melanomas. Vemurafenib targets this altered form of BRAF and has been approved to treat metastatic or inoperable melanomas. A third approach is looking for abnormal chromosomes present in cancer cells but not in normal cells. Chromosomal abnormalities can result in gene fusion, wherein parts of two different genes are incorporated as one gene. These fusion proteins can increase cancer development. Imatinib mesylate is one example of a therapy that targets the BCR-ABL1 fusion protein responsible for the growth of some leukemic cells.
Targeted therapies do have limitations. One limitation is that cancers that do not have sufficient quantities of the specific molecular targets will not respond to targeted therapy. Another limitation involves cancer cells mutating and becoming drug resistant. Finally, yet another limitation of targeted therapy is the difficulty of developing drugs for some identified targets.
Pathophysiology
Normal Cell-Growth Regulation
Genetic Control Over Cell Division
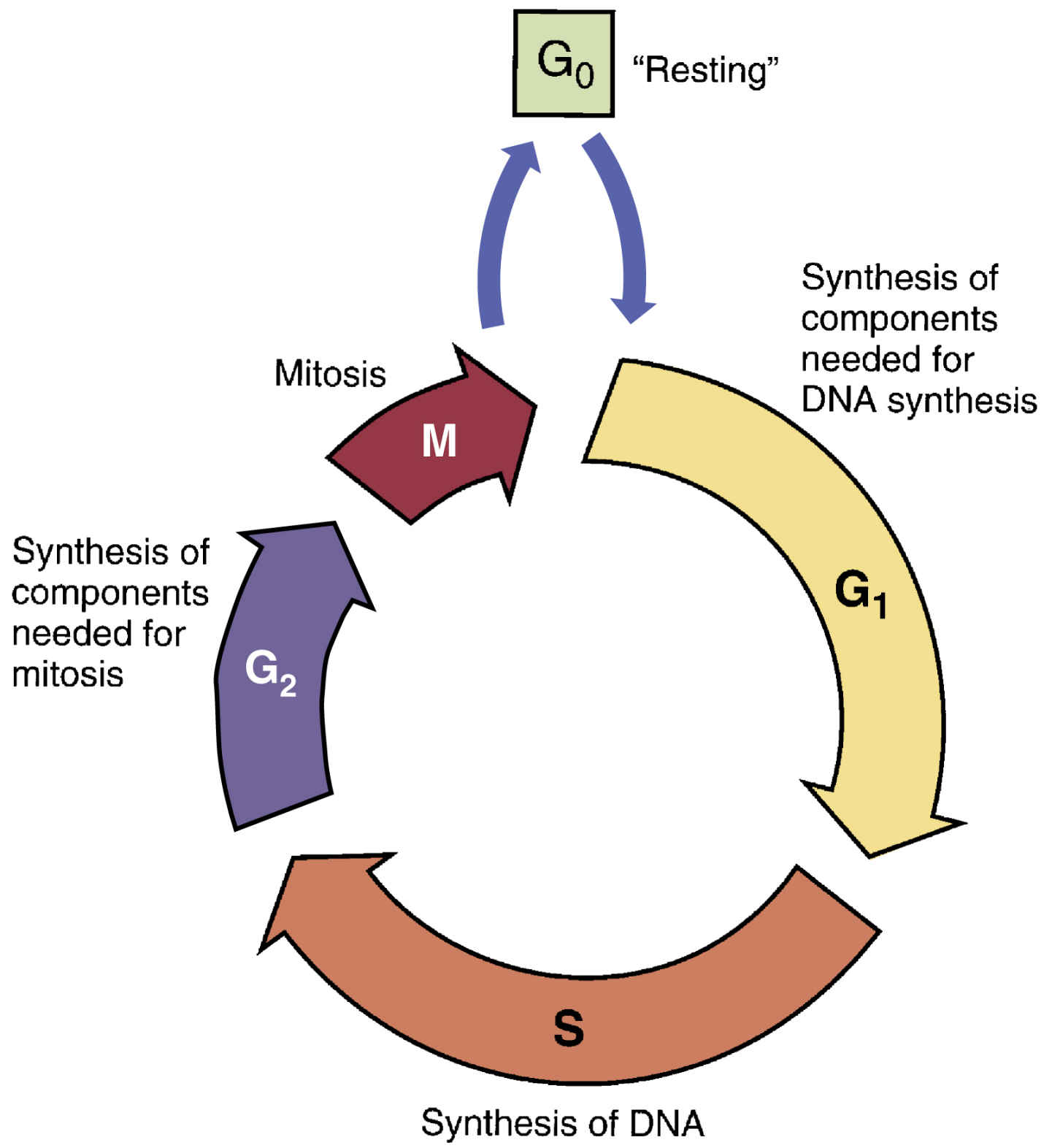
Some cancers are inherited, but others develop when genetic material becomes deranged or mutated. For malignancy to develop, cells must undergo more than one mutation; for this reason, malignancy may take years to develop. As depicted in Fig. 33.1, most if not all cells progress through the five phases of the cell cycle, which comprise a presynthesis phase (G1), synthesis phase (S), postsynthesis phase (G2), mitosis phase (M), and resting phase (G0). Cells grow in size during the G1 phase, DNA is copied during the S phase, the cell prepares to divide in the G2 phase, and the cell divides in the M phase. A cell may remain in the G0 phase for days to years, and under appropriate conditions, it will start participating in the cell cycle. Before entering a cell cycle, cells must pass through various cellular checkpoints, the function of which is to determine whether a cell has completed the necessary steps prior to progressing in the cell cycle. There are three major checkpoints in the cell cycle: one between the G1 and S phases, one between G2 and mitosis, and one during mitosis. A cell undergoes apoptosis if it is found to be defective at any one point during the cell cycle.
Cellular communication and signal transduction are mechanisms in which cell-to-cell communication occurs locally or remotely by signal molecules sent to maintain homeostasis; to regulate growth and division; to develop and organize into tissues; and to coordinate cellular functions. Three primary means of cellular communications include direct contact with plasma membrane-bound receptors, remote contact with secreted signal molecules, and direct contact by way of gap junctions. Secreted signal molecules affect intercellular communication in a variety of ways, primarily by mechanisms that are contact dependent, paracrine, hormonal, and neurohormonal and also through neurotransmitters. Cells respond to external stimuli by activating a variety of signal transduction pathways, or signaling cascades. Fig. 33.2 illustrates a segment of a cell with one interconnecting signal transduction pathway that, when activated, leads to gene activation for those proteins that promote cell division. This pathway can be activated by (1) growth factors binding to cell receptors; (2) the interaction of certain drugs with the cell plasma membrane; (3) the presence of adhesion proteins; (4) changes in ion movement, especially sodium and calcium; (5) ligand binding; and (6) other cell-to-cell interactions. The signaling molecules in turn activate protein kinase pathways, increasing tyrosine kinase inside the cell.
A tyrosine kinase (TK) is an enzyme that activates other substances by adding a phosphate group (PO4) to them, a process known as phosphorylation. Some families of TKs are unique to a cell type, whereas others may be present only in cancer cells that express a specific gene mutation. Regardless of how a pro–cell-division signal transduction pathway is activated, the result is an increase in TK levels that propagate the signal by activating many different transcription factors within the pathway.
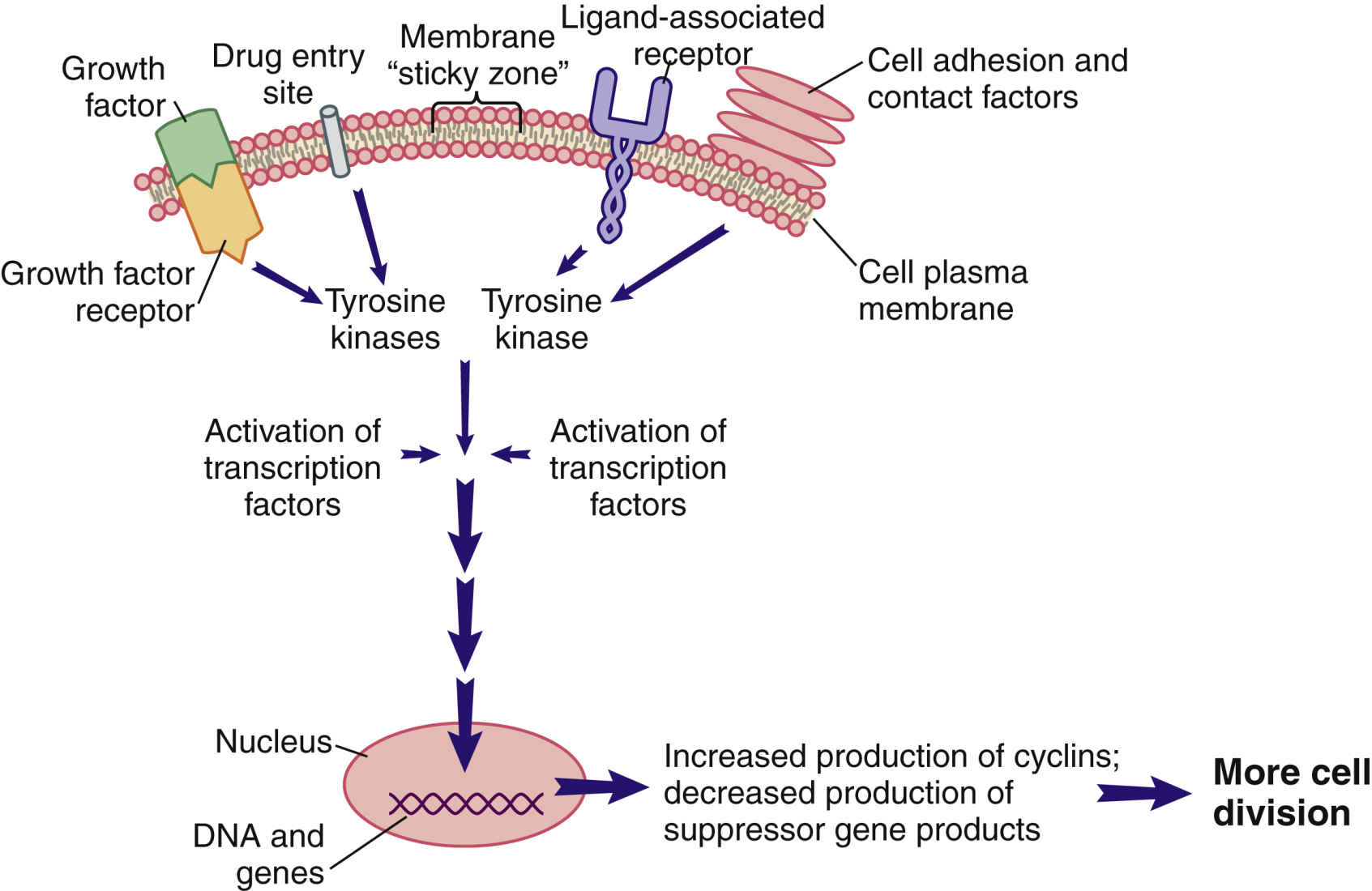
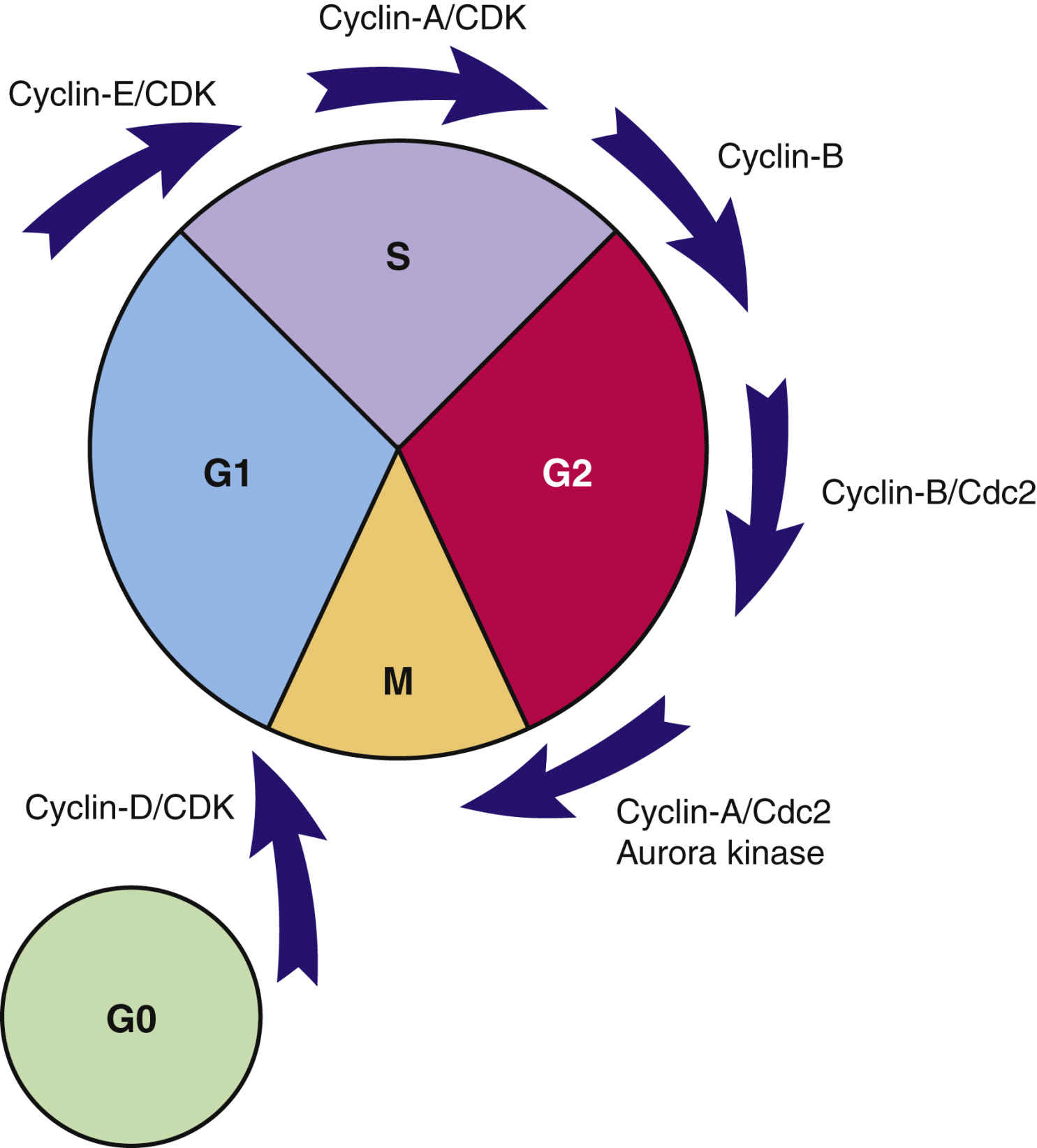
Transcription factors are substances that enter the nucleus and signal the cell to begin mitosis. Many different types of substances serve as transcription factors. The overall response is a greater expression of oncogene products (cyclins) that promote cell division and reduced expression of suppressor gene products that inhibit cell division. When the cell responds to these mitotic signals, indicating cell division is needed and resources are adequate, the cell leaves the G0 phase and enters the G1 phase of the cell cycle. Once the cell enters the G1 phase, the presence of specific cyclins determines whether it can progress to the next phase (Fig. 33.3).
Cyclins are a family of proteins that stimulate the cell to move through the cell cycle. Cyclins are the products of oncogenes, and most are activated when a phosphorous group is added to the cyclin chemical structure. (Removal of a phosphorous molecule from a cyclin [dephosphorylation] inhibits its activity.) Kinases that activate cyclins are called cyclin-dependent kinases (CDKs). CDKs combine with cyclins to form complexes that initiate cell mechanisms to complete cell division. The amount and type of cyclins and CDKs present in the cell during cell division vary by the phase of the cycle. These differences in types of cyclins and CDKs determine when or if a particular cell moves from one phase to the next. Of the cyclins identified, those in group D are the most studied and understood.
A common signal for entering and starting the cell cycle at G1 is the combining of a cyclin-D with the appropriate CDK to form a cyclin-D/CDK complex (see Fig. 33.3). Movement of the cell from G1 into other phases of the cell cycle is regulated by the continued presence of pro–cell-division transcription factors that promote DNA transcription and increased synthesis of specific pro–cell-division cyclins and CDKs.
In normal cells, the oncogenes that produce cyclins are carefully regulated by suppressor gene products so that cell division only occurs when it is needed and to the degree that it is needed. Proteins synthesized by suppressor genes determine how much oncogene expression is needed to allow cell division to occur without excessive cell division. Such control is exemplified by normal wound repair. For example, when a person falls and scrapes skin from the knee, the skin cells at the edge of the wound are signaled to divide and fill in the gap. When the wound area is closed, cell division normally stops; thereby, uncontrolled cell division does not occur.
When cell division is not needed, external signals—such as growth factor inhibitors and the surrounding of a cell plasma membrane with other cells—send signals that are inhibitory to the pro–cell-division signal transduction pathway (Fig. 33.4). The result of this inhibition leads to low levels of TKs and reduced levels of pro–cell-division transcription factors. In this scenario, suppressor gene activity is increased, which results in production of more suppressor gene products that inhibit the synthesis of cyclins and CDKs by oncogenes. Even though many different types of suppressor genes are present, certain suppressor genes become active depending on the cell type. For example, the BRCA1 suppressor gene appears to be most active in suppressing excessive cell division in breast, ovary, and genitourinary tract tissues. Without suppressor gene products, oncogenes would be overexpressed continually, leading to uncontrolled and unneeded cell division.
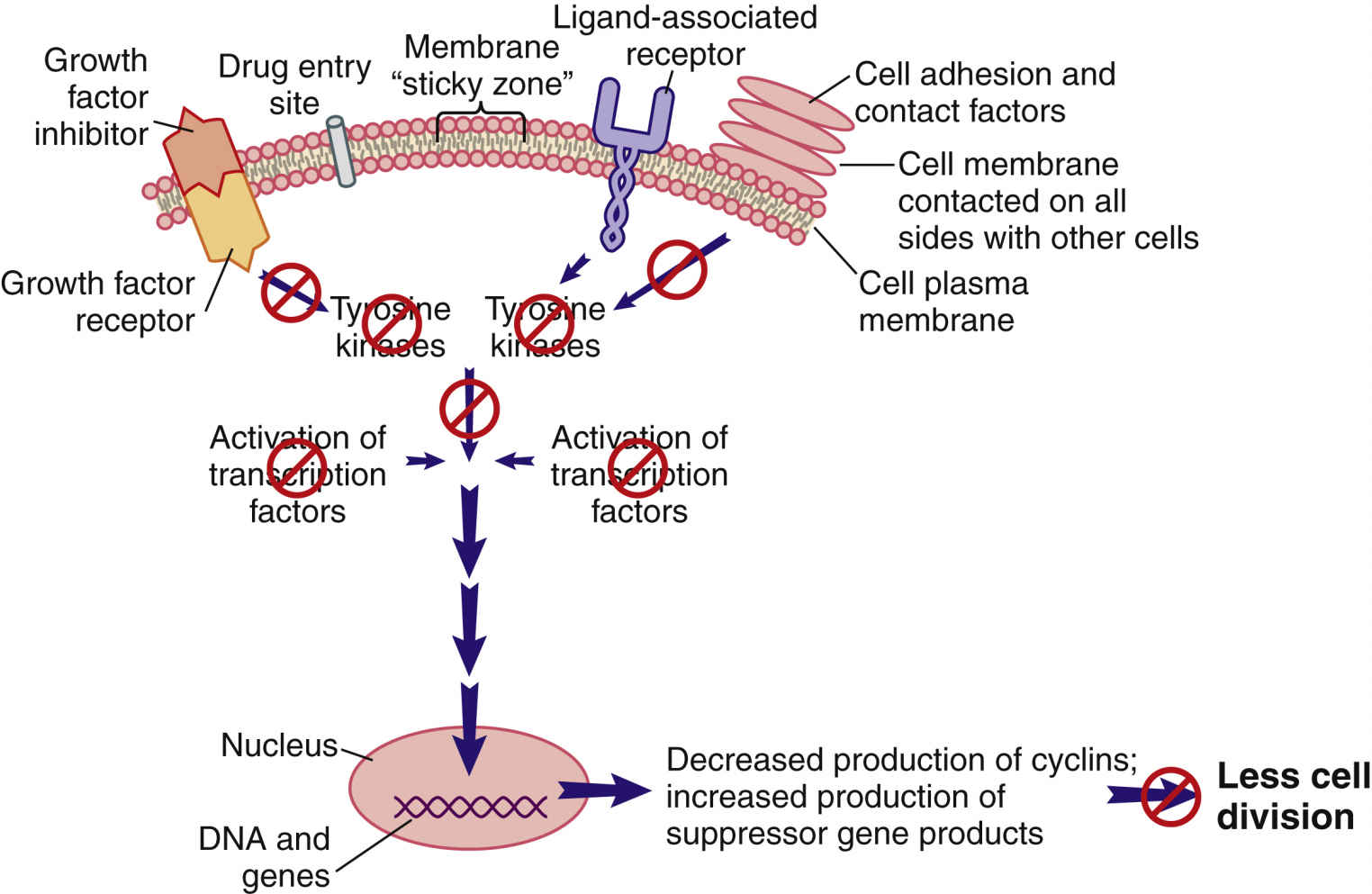
FIG. 33.4 External signals that inhibit the signal transduction pathway, resulting in greatly reduced cell division.
Internal cell conditions, such as poor cell nutrition and reduced energy stores, can inappropriately trigger the activation of suppressor genes to disrupt the pro–cell-division signal transduction pathway, even when external conditions indicate a need for cell division (Fig. 33.5).
Genetic Control Over Cell Death
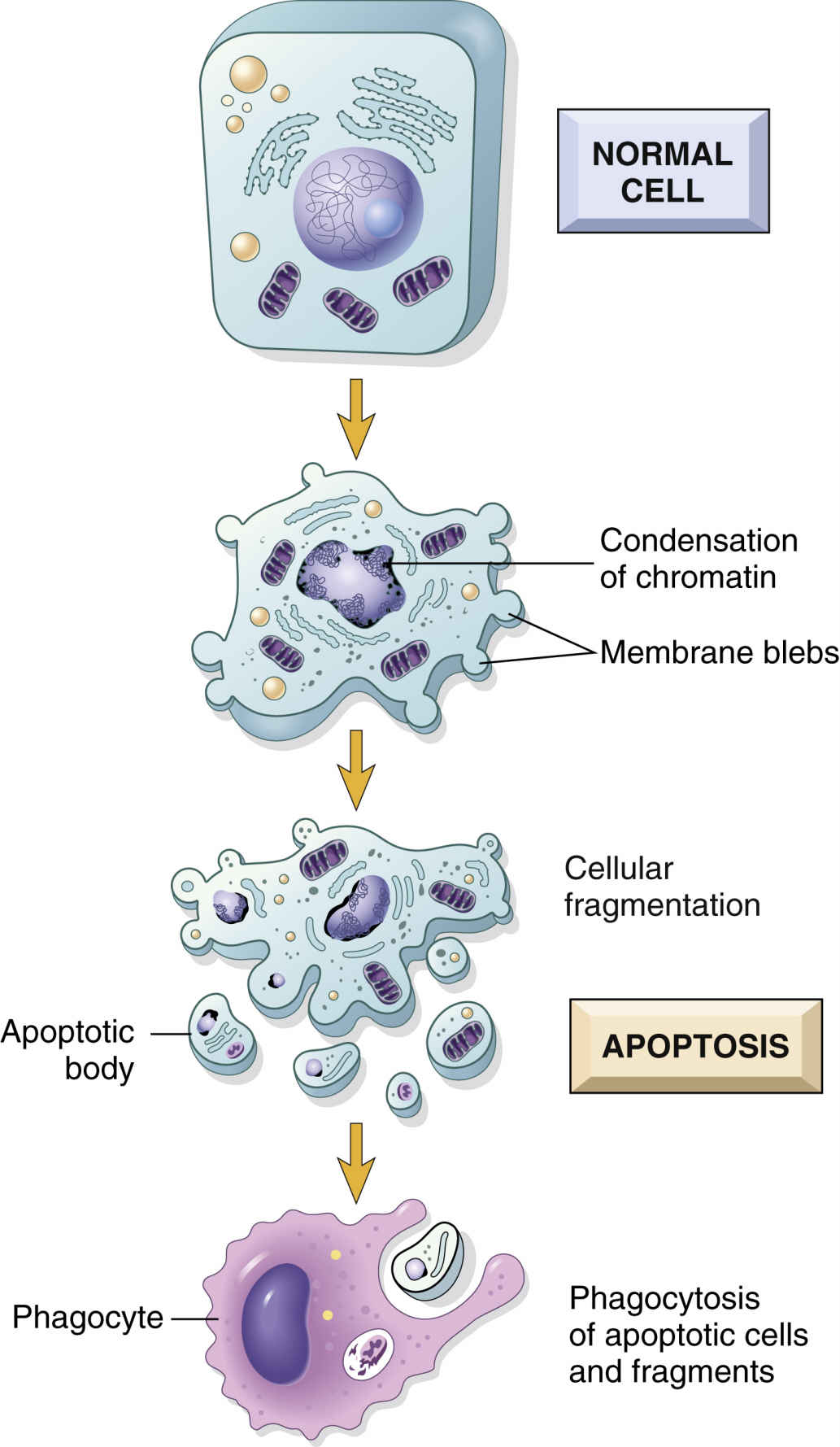
As cells age, their function declines; function in damaged cells declines even earlier. In normal tissues that are capable of cell division, damaged cells and older cells respond to apoptosis (Fig. 33.6), or programmed cell death. Apoptosis is a natural way of removing aged cells from the body to ensure that tissues and organs contain healthy and optimally functional cells. Through genetic control, apoptosis prevents healthy, functional cells from self-destructing prematurely, and it eliminates older or damaged cells, thus preventing an overpopulation of cells that could reduce organ efficiency. Maintenance of optimally functional organs depends on a balance of cell division with apoptosis.
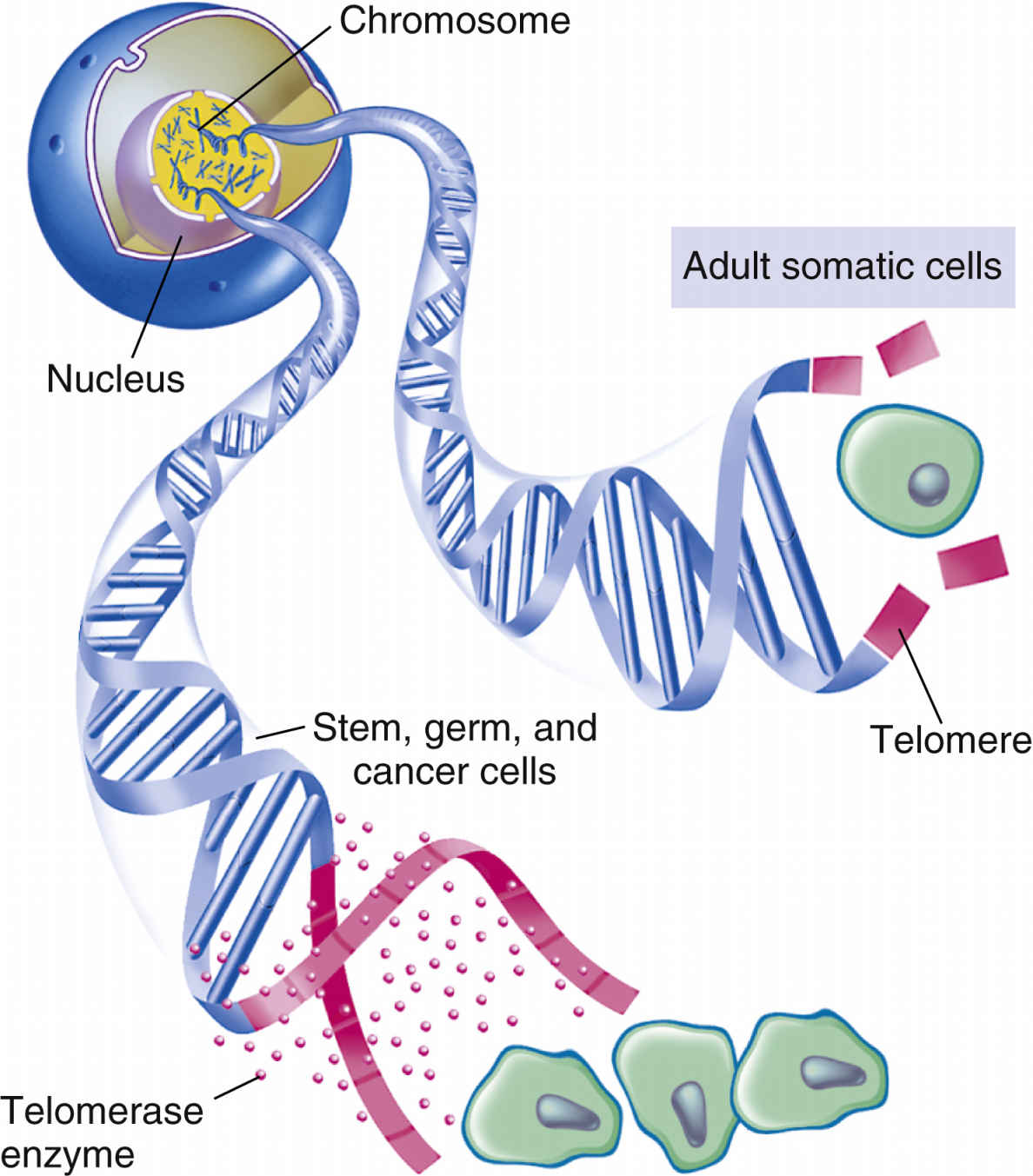
Telomeres are specialized, repetitive DNA sequences located at the ends of chromosomes. The signals for apoptosis may begin with the loss of these telomeres in aging cells. Telomeric DNA shortens with each cell division, eventually halting the dividing of cells (Fig. 33.7). When the cell has undergone its lifespan’s worth of cell divisions, the telomeric DNA that capped the chromosomes disappears, allowing the ends to unravel and fragment. These processes then trigger genetic and other intracellular signals for self-destruction through the action of autoenzymes of caspases. Activated caspases degrade the cell’s internal structures, causing the plasma membrane to lyse the cell into small fragments that are then removed by the body’s defense systems.
Growth Regulation and Cancer
Loss of Genetic Control of Cell Growth
Normal cells become cancer cells when external or internal conditions lead to gene damage. Cancer cells develop because of damaged suppressor genes, overproduction of cyclins and CDKs, or reactivation of telomeres that allow damaged cells prolonged life.
Excessive cell division from gene damage and mutations appears to be self-perpetuating, which leads to further gene mutations that:
• Allow one or more specific pro–cell-division signal transduction pathways to become more active.
• Allow greater expression of cancer cell membrane receptors that trigger pro–cell-division signal transduction pathways.
• Increase or amplify the production of specific TKs and transcription factors.
Loss of Apoptosis
The inability of cells to undergo apoptosis allows aged, damaged cells to undergo uncontrolled proliferation, resulting in tumor development, progression of tumors to cancer, and treatment resistance. A cell’s resistance to apoptosis is multifactorial and involves expression of many antiapoptotic molecules. Mutations that occur in suppressor genes as a result of DNA damage inactivate the suppressor genes, preventing them from controlling oncogene activity. Some suppressor genes, such as the TP53 gene, also regulate apoptosis. Inactivation of suppressor genes that regulate apoptosis makes cancer cells unresponsive to apoptotic signals. These cancer cells are now resistant to natural cell death, a feature known as cellular immortality. The lack of regulation for cell division and the loss of apoptosis result in cancer cells having no balance between cell division and apoptosis. This disequilibrium favors continuous cancer cell division.

Autophagy and Resistance to Cancer
Intracellular homeostasis is maintained through autophagy, a process that involves lysosomal degradation and recycling of unnecessary or damaged cellular components. Autophagy can prevent cellular damage caused by chemotherapeutics by removing dysfunctional cellular parts and eliminating cellular stress. Autophagy has also been suggested as an important mechanism in the survival of tumor cells against apoptosis when exposed to certain anticancer drugs, leading to chemoresistance.

Targeted Therapy Drugs
Once a cell receives signals, the signal is relayed to other cells by a series of biochemical reactions. In cancer cells, the cells are stimulated to divide continuously without being prompted by external growth factors. Signal transduction inhibitors (STIs) block signals to cancerous cells by blocking signals passed from one molecule to another. They work at sites that are on the cell surface, at the intracellular level, or in the extracellular domain. Blocking various signals can affect cell division and can ultimately cause cell death. Targeted cancer therapies are STIs that block signal transduction among specific molecules, which blocks the growth and spread of cancerous cells. Drugs that target specific cells inhibit cancer cell division by blocking cancer membrane receptors and TK activities, interfering with signal transduction, stimulating immune system attacks on cancer cells, or inducing cells to undergo apoptosis. Targeted drugs may be used as monotherapy (a single agent), in combination with traditional chemotherapy, and with radiotherapy.
Targeted therapies are more selective for specific molecular targets than cytotoxic anticancer drugs, thus they are able to kill cancer cells with less damage to normal cells compared with chemotherapy. They are the cornerstone of precision medicine that uses a person’s genes and proteins to prevent, diagnose, and treat cancer. Targeted therapies are sometimes known as molecular targeted drugs and precision medicines. Most targeted therapies consist of either small molecules or monoclonal antibodies (MAbs). Targeted therapies include angiogenesis inhibitors, epidermal growth factor receptor (EGFR)–TK inhibitors, BCR-ABL1 tyrosine kinase inhibitors, MAbs, and proteasome inhibitors. Small-molecule drugs are not antibodies. Most generic small-molecule drugs have –nib or –mib suffixes, such as imatinib and bortezomib. Monoclonal antibodies have –mab suffixes, such as rituximab. Table 33.1 identifies other suffixes common to small-molecule drugs and MAbs. Small-molecule drugs are usually administered orally, whereas MAbs are given intravenously. All targeted therapies are categorized as high-alert medications.
Targeted therapies differ from standard chemotherapy in several ways. Targeted therapies (1) act on specific molecular targets associated with cancer, (2) are deliberately designed to interact with a specific target, and (3) often are cytostatic by blocking tumor cell proliferation.
However, targeted therapy does have drawbacks. Cancer cells can become resistant, and drugs for some targets are difficult to develop. Side effects and risks associated with targeted therapy include diarrhea, hepatitis, thrombus formation, poor wound healing, hypertension, fatigue, stomatitis, and skin changes, among others.
The rapid identification of specific cancer cell targets in recent years has led to an increased development of targeted therapies. Management of patient issues related to targeted therapies is an evolving area of study. With many targeted therapies being new to the market, costs can be prohibitive.
TABLE 33.1
Suffixes of Targeted Cancer Therapies
| Suffix | Meaning |
| -mab | A monoclonal antibody |
| -momab | A monoclonal antibody composed of only murine (mouse) proteins |
| -imab | A monoclonal antibody composed of more human proteins (>60%) than murine proteins (∼30%) |
| -zumab | A monoclonal antibody composed of mostly human proteins (95% or more) and only a few murine proteins (<5%) |
| -umab | A monoclonal antibody composed of only human proteins and no murine proteins |
| -nib | A tyrosine kinase inhibitor |
| -mib | A proteasome inhibitor |
Small-Molecule Compounds
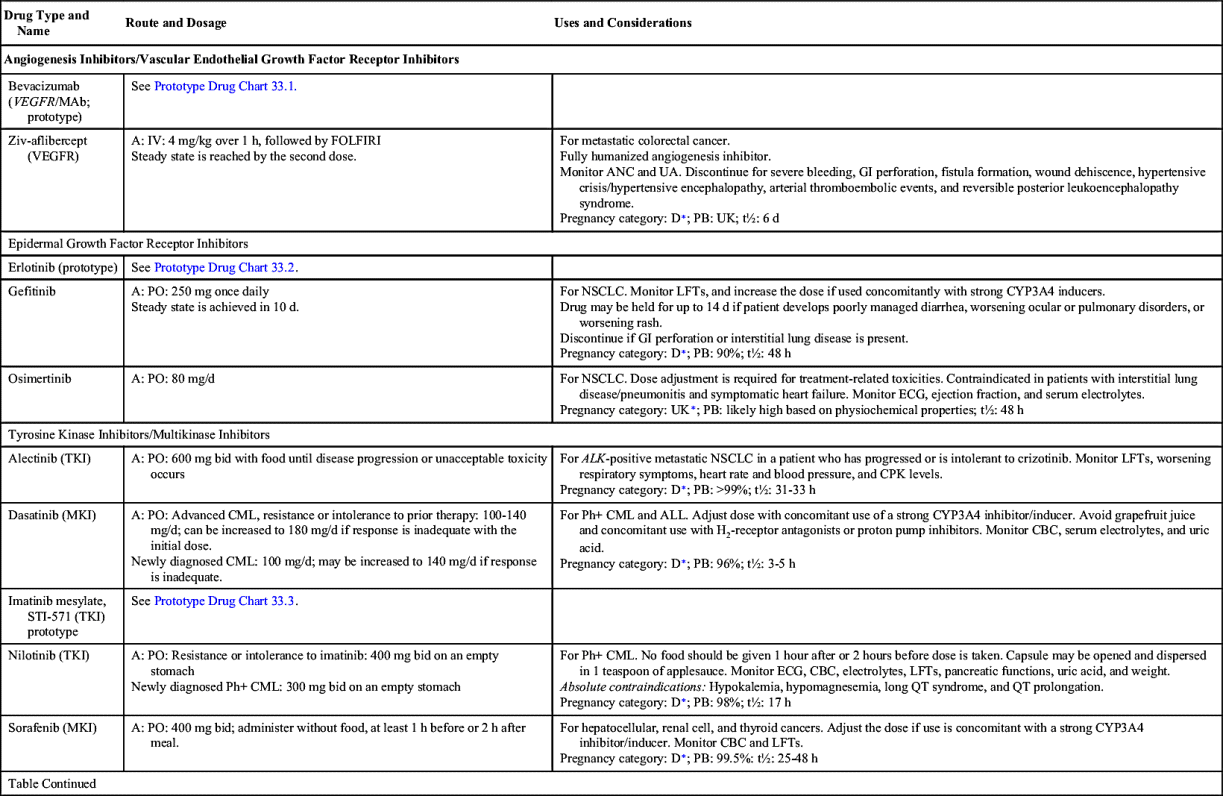
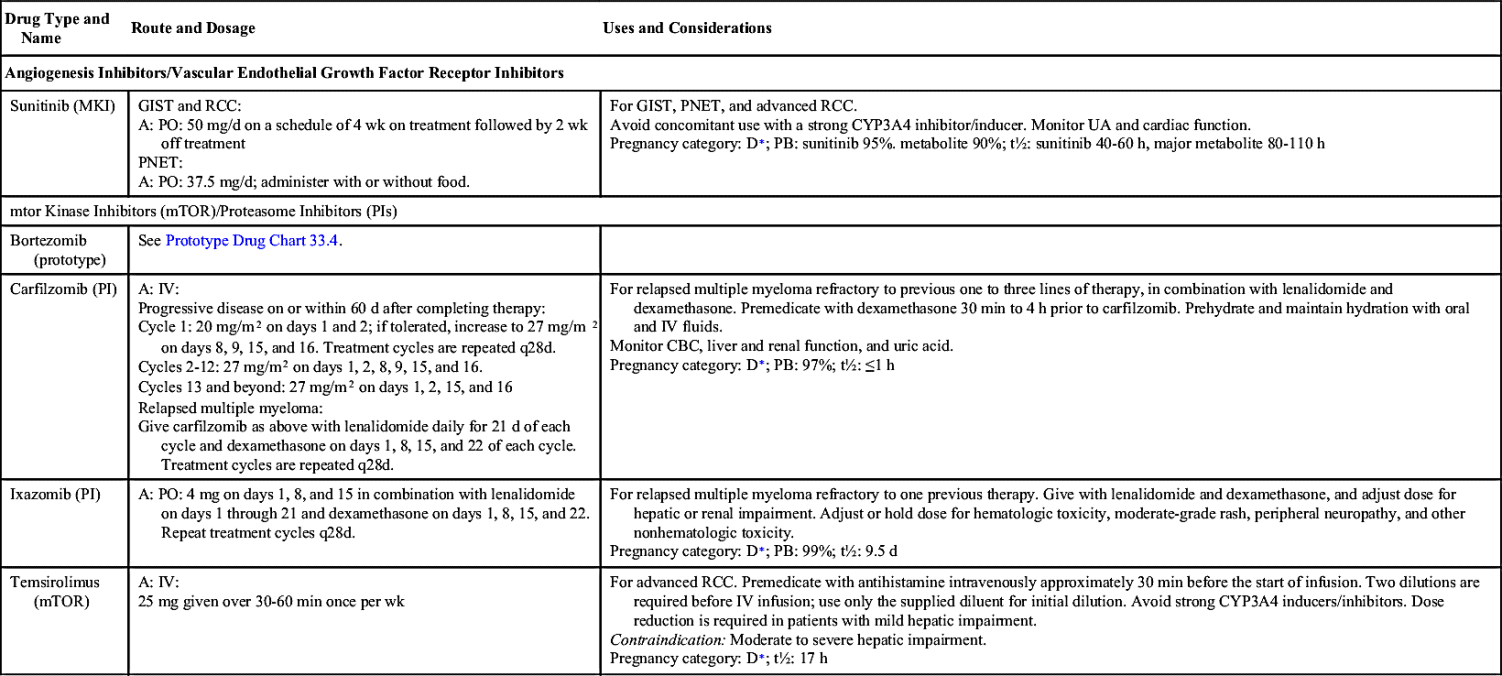
Small-molecule compounds are one of the two main types of targeted therapy. Like most other types of drugs, they are chemicals that are small enough to have an intracellular effect, targeting the internal structures of cells. Small-molecule drugs are classified according to their actions; these include inhibiting enzymes, inducing apoptosis, and inhibiting formation of new vasculatures (angiogenesis). Table 33.2 lists the small-molecule inhibitors (SMIs) and their dosages, routes, uses, and considerations.
Angiogenesis Inhibitors/Vascular Endothelial Growth Factor Receptor Inhibitors
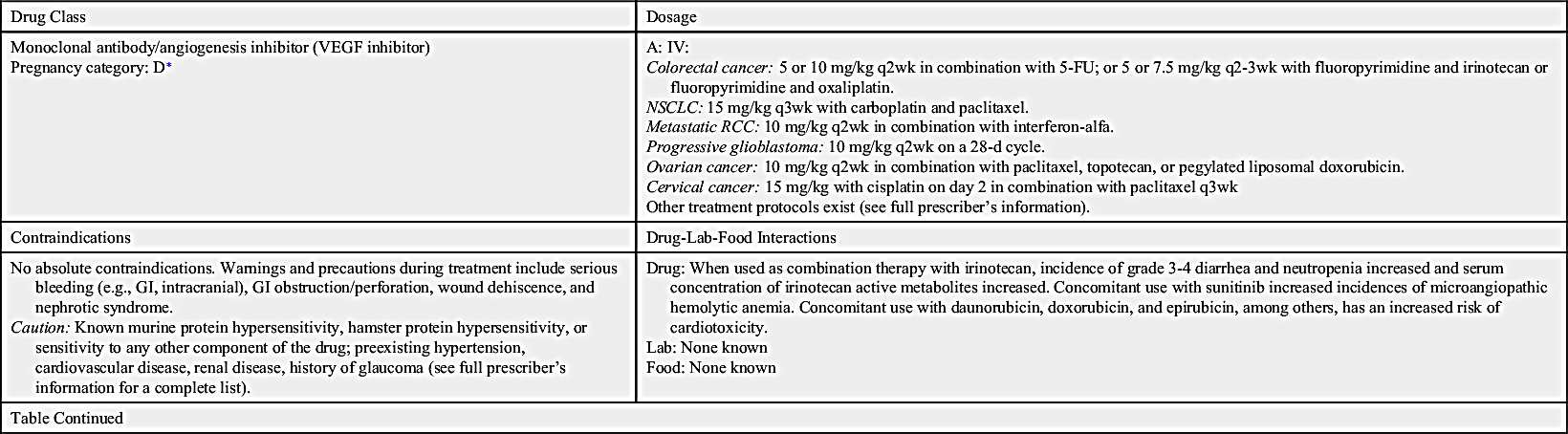
Angiogenesis inhibitors prevent new blood vessels from forming, which is required for tumors to grow. Some angiogenesis inhibitors interfere with the action of vascular endothelial growth factor (VEGF), a substance that stimulates new blood vessel formation. Others prevent the formation of platelet-derived growth factor (PDGF) that is intricate in the formation of new blood vessels and in the growth of preexisting blood vessels. Some angiogenesis inhibitors are also considered monoclonal antibodies (MAbs), such as bevacizumab. Prototype Drug Chart 33.1 lists specific drug information for bevacizumab.
Ziv-aflibercept
Ziv-aflibercept is a fully humanized recombinant fusion protein that inhibits angiogenesis. It is indicated in combination with 5-fluorouracil (5-FU), leucovorin, and irinotecan (FOLFIRI) for the treatment of metastatic colorectal cancer that is resistant to or has progressed after receiving an oxaliplatin-containing regimen.
Pharmacokinetics
Ziv-aflibercept is administered by intravenous (IV) infusion. Its elimination half-life is approximately 6 days, which appears to be dose dependent. The steady state is also dose dependent, with the average steady state reached by the second dose in a 2-week regimen. The metabolic and elimination pathways are unknown. However, free ziv-aflibercept does not seem to be affected by mild to moderate hepatic or renal impairment.
Pharmacodynamics
Ziv-aflibercept is an angiogenesis inhibitor that acts as a soluble receptor to bind vascular endothelial growth factors A and B and placental growth factors 1 and 2, which prevents other native receptors from binding. This action can result in decreased neovascularization and decreased vascular permeability.
Side effects and adverse reactions
Severe bleeding, including gastrointestinal (GI) bleeding, intractable bleeding, and pulmonary hemorrhage, has been noted. GI perforations, some fatal, have also been reported. Other adverse events include compromised wound healing and / or wound dehiscence, reversible posterior leukoencephalopathy syndrome (RPLS), hypertensive crisis, nephrotic syndrome, or thrombotic microangiopathy. Common side effects include diarrhea, dizziness, asthenia, weight loss, and dehydration.
Drug interactions
No interactions have been reported.
Epidermal Growth Factor Receptor Inhibitors
The largest class of targeted therapy that attacks one particular molecular target is epidermal growth factor receptor (EGFR) inhibitor. These receptors are overexpressed, dysregulated, or mutated in many epithelial cancers. EGFR expression has been associated with poor prognosis, metastasis, chemotherapy resistance, hormonal therapy, and radiation therapy. The activation of EGFRs is important in cancer cell growth and proliferation. Most EGFR inhibitors also inhibit tyrosine kinase (TK) indirectly. However, two other classes of EGFR inhibitors target EGFR extracellularly (MAbs) and also target the receptor catalytic domain of EGFR (TK inhibitors). These two classes are further discussed under the headings Monoclonal Antibodies and Tyrosine Kinase Inhibitors and Multikinase Inhibitors. The EGFR inhibitors that inhibit TK indirectly include erlotinib, gefitinib, and osimertinib. As shown in Fig. 33.2, the growth factor receptors on the cell membrane can activate TKs, which then turn on signal transduction pathways that promote cell division. The EGFR inhibitors bind to different areas of the EGFR, blocking its activity so that it cannot activate TK. As a result, the downstream signal transduction pathway for promotion of cell division is inhibited, and cell proliferation is severely limited. Table 33.2 lists the EGFR inhibitors and their dosages, routes, uses, and considerations. Prototype Drug Chart 33.2 lists the specific drug information for erlotinib.


Gefitinib
Gefitinib is a synthetic anilinoquinazoline that selectively inhibits the epidermal growth factor receptor tyrosine kinase (EGFR-TK). It is most commonly used in the management of locally or advanced metastatic non–small cell lung cancer (NSCLC).
Pharmacokinetics
Gefitinib is taken orally and is absorbed slowly in the GI tract, with 60% reaching systemic circulation. Peak plasma level occurs 3 to 7 hours after dosing. No significant alteration in its bioavailability was noted when gefitinib was taken with food. It is metabolized mainly by the hepatic system primarily through the CYP3A4 enzyme. Gefitinib is primarily excreted in the feces, mostly in the form of its metabolites. The elimination half-life is approximately 48 hours.

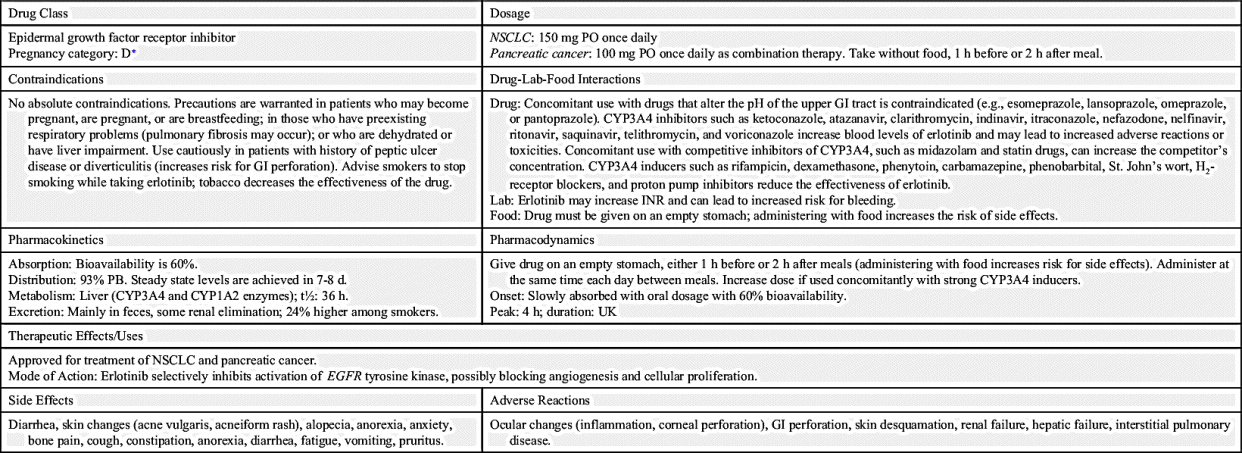
Pharmacodynamics
Gefitinib reversibly binds to the adenosine triphosphate (ATP) binding site and completely inhibits autophosphorylation by EGFR-TK; EGFR-TK functions as a mediator of cell growth, differentiation, and death. This action results in blockage of downstream EGFR-TK–mediated signal transduction pathways, cell cycle arrest, and inhibition of angiogenesis.
Side effects and adverse reactions
Common side effects include skin reactions, diarrhea, anorexia, vomiting, and elevated transaminases. Gefitinib can cause conjunctivitis and abnormal eyelash growth, and rash occurs in about 47% of patients. Patients can also experience acne, pruritus, and nail disorders. Interstitial lung disease and GI perforation were also reported.
Drug interactions
Gefitinib is extensively metabolized by CYP3A4. Other drugs metabolized by the hepatic system can affect the concentrations of gefitinib and other drugs. Drugs that inhibit CYP3A4, increasing the gefitinib level, include thioridazine, amiodarone, bupropion, diphenhydramine, promethazine, and metoclopramide. Other CYP3A4 inhibitors, such as venlafaxine, may decrease the levels of gefitinib. Gefitinib can increase the concentration of other drugs that have a narrow therapeutic index, such as amitriptyline, warfarin, and eliglustat. Patients taking warfarin concomitantly with gefitinib had an elevated international normalized ratio (INR) and/or hemorrhage. CYP3A4 inducers—such as acetaminophen, butalbital, caffeine, and aspirin—can increase gefitinib metabolism, thereby decreasing concentrations of the drug. When given concomitantly with gefitinib, drugs that decrease gastric pH (e.g., omeprazole and lansoprazole) can lower plasma concentrations of gefitinib.
Osimertinib
Osimertinib is one of the newest FDA-approved EGFR-TK inhibitors for T790M-mutation positive NSCLC. Osimertinib was approved in November 2015.
Pharmacokinetics
Osimertinib is taken orally, and absorption is dose dependent. High-fat, high-calorie meals increase the absorption rate by 14%. It is metabolized primarily by the hepatic system through the CYP3A isoenzyme and by oxidation and dealkylation. Osimertinib is primarily excreted in the feces and to a lesser extent in the urine. The elimination half-life is approximately 48 hours.
Pharmacodynamics
Osimertinib is a CYP3A4 and CYP1A2 inducer that irreversibly binds to certain mutant forms of EGFR (T790M, L858R, and exon 19 deletion). In vitro it exhibited inhibition of ERBB2, ERBB3, ERBB4, ACK1, and BLK receptors.
Side effects and adverse reactions
Common side effects include pancytopenia, rash, dry skin, anorexia, constipation, hyponatremia, nausea, pruritus, fatigue, cough, back pain, and stomatitis. Other side effects include eye disorders and cephalgia. Adverse reactions include venous thromboembolism, interstitial lung disease, pneumonitis, increased QTc, and cardiomyopathy.
Drug interactions
Osimertinib is a CYP3A inducer. Drugs metabolized by the liver can affect their concentrations and the osimertinib plasma level. Osimertinib can cause a prolonged QT interval. Other drugs that can prolong QT intervals, causing torsade de pointes, should be avoided or monitored closely when used concomitantly with osimertinib (e.g., thioridazine, beta agonists, chlorpromazine, ofloxacin, desipramine, moxifloxacin, metronidazole, promethazine, and trimethoprim or sulfamethoxazole-trimethoprim). CYP3A inhibitors such as aldesleukin, alfentanil, and ethanol can alter osimertinib concentration. Osimertinib can alter plasma concentration of other drugs, which can worsen adverse reactions and can alter the efficacy of drugs such as acetaminophen, amlodipine, amitriptyline, and alprazolam.
 Nursing Process: Patient-Centered Collaborative Care
Nursing Process: Patient-Centered Collaborative CareAngiogenesis Inhibitors, Vascular Endothelial Growth Factor Receptor Inhibitors, and Epidermal Growth Factor Receptor Inhibitors
Assessment
• To avoid drug interactions, obtain a detailed medication history that includes current prescriptions, over-the-counter (OTC) medicines, antacids, dietary supplements, vitamins, and herbal supplements.
• Obtain a list of all drug and food allergies.
• Obtain baseline information about the patient’s physical status that includes height, weight, vital signs, cardiopulmonary assessment, intake and output, skin assessment, nutritional status, and any underlying diseases.
• Obtain baseline laboratory values (complete blood count [CBC], uric acid, chemistry panel) before and during treatment.
• Assess baseline results of pulmonary function tests, chest radiographs, electrocardiographs (ECGs), and renal and liver function studies.
• Assess patient’s and caregiver’s current level of comprehension related to the therapeutic regimen.
Nursing Diagnoses
• Skin Integrity, Risk for Impaired
• Infection, Risk for related to bone marrow suppression
• Knowledge, Deficient related to the targeted therapy regimen
• Injury, Risk for related to toxic effects of targeted therapy
• Sensory Perception, Risk for Disturbed Visual
Planning
• Skin will maintain its integrity.
• The patient will remain free from infection.
• The patient and family will verbalize understanding of targeted therapy as part of an anticancer treatment regimen.
• The patient and family will verbalize strategies to minimize risks related to targeted therapy–related side effects.
• The patient and family will demonstrate understanding of the importance of reporting targeted therapy–related side effects and adverse reactions.
• The patient’s side effects will be managed to a tolerable level and are not life threatening.
Nursing Interventions
• Examine the patient’s skin closely at each visit for the presence of erythema, rash, peeling, or blister formation; rate the severity of dermatologic reactions.
• Monitor for any evidence of infection, such as fever, chills, leukocytosis or leukopenia, and neutropenia.
• Assess for evidence of thromboembolic events.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


