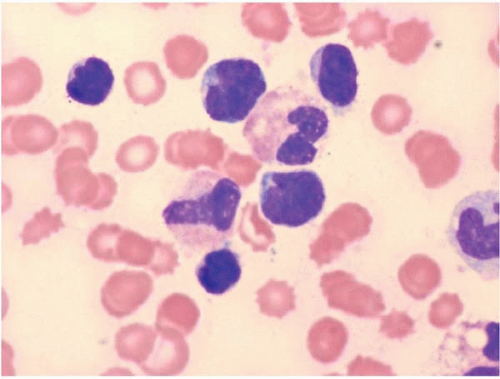
Adult T-Cell Leukemia’Lymphoma
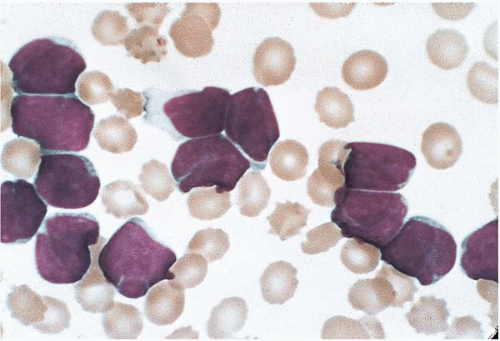
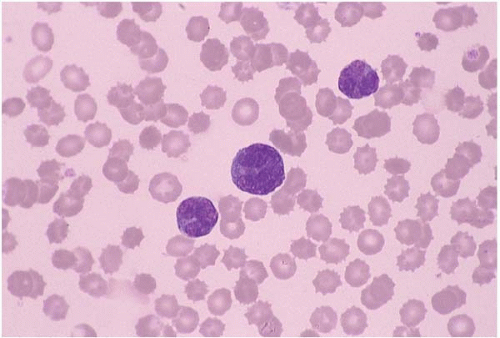
Adult T-cell leukemia’lymphoma (ATLL) arises as a consequence of human lymphotropic virus I (HTLV-I) infection (
Figs. 26.2 and
26.3) (
28,
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75). The clinical course of the HTLV-I infection ranges from smoldering disease to chronic disease, lymphoma, and leukemia.
Patients with ATLL may present with disease involving lymph nodes, liver, spleen, peripheral blood, and bone marrow. Skin lesions, hypercalcemia, and opportunistic infections may be present. ATLL occurs both in children and adults. The clinical course is usually aggressive, and the prognosis is poor. Spontaneous remission has been reported but is usually transient.
Laboratory studies may show anemia, neutrophilia, thrombocytopenia, monocytosis, eosinophilia, and’or basophilia. The absolute lymphocyte count is variable.
Peripheral blood and bone marrow smears show abnormal lymphocytes. These are typically a heterogeneous population of cells, consisting of a range of small and large cells. Nuclei range from small and irregular to large and prominently lobated. Nuclear hypersegmentation has prompted the term “flower cells.” Nucleoli may be present. The cytoplasm is scant to moderately abundant and basophilic and may contain inclusions, vacuoles, and’or granules. Tartrate-resistant acid phosphatase activity may be present.
Histologic sections of the bone marrow show abnormal lymphocytes distributed as a patchy or diffuse interstitial infiltrate.
Many morphologic variants are recognized. ATLL may resemble Burkitt lymphoma’leukemia, lymphoblastic lymphoma, T-cell chronic lymphocytic leukemia, anaplastic large cell lymphoma, mycosis fungoides’Sézary syndrome, and angioimmunoblastic T-cell lymphoma.
Other findings include hemophagocytic syndrome, which may obscure the underlying disease, and osteitis fibrosa cystica, owing to tumor cell secretion of parathormone-related protein.
Immunophenotyping usually shows expression of CD2, CD3, CD4, CD7, CD25, and TCR α-β. CD4 and CD25 are consistently expressed. Compared to normal T-cells, expression of pan-T-cell antigens (CD2, CD3, CD5, CD7) may be reduced or absent. CD3, in particular, shows low expression. Some cases express both CD4 and CD8, CD8 only, or neither CD4 nor CD8. In CD8+ cases, granzyme B and TIA-1 (but not perforin) may be expressed. CD20, CD30, CD56, epithelial membrane antigen, and S100 protein expression have been reported.
Genetic studies have shown various clonal karyotypic anomalies, especially del(6q), and rearrangements and deletions involving the TRC genes. Clonal integration of human T-cell lymphotropic virus type 1 (HTLV-1) proviral DNA into tumor cells is consistently found.
ATLL has been reported with disseminated xanthogranulomas, myelodysplastic syndrome, acute myeloid leukemia, and various B-cell neoplasms.
The differential diagnosis includes other T-cell neoplasms, clonal B-cell and NK-cell disorders, and acute leukemia.
Anaplastic Large T-Cell Lymphoma
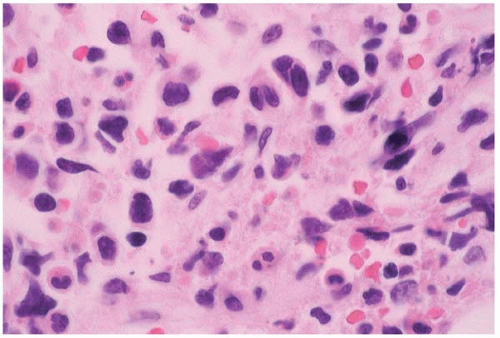
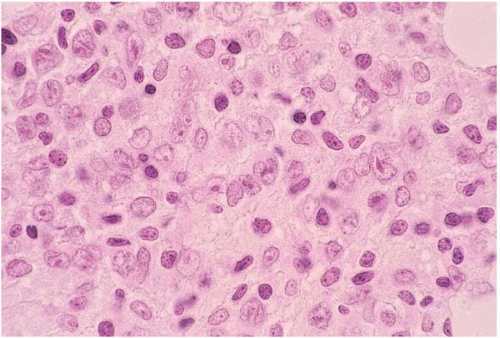
Anaplastic large cell lymphoma (ALCL) arises de novo; as a complication of immunosuppressed states, including human immunodeficiency virus infection and solid organ transplantation; and as a transformation of mycosis fungoides’Sézary syndrome and T-cell prolymphocytic leukemia (
Figs. 26.4 and
26.5) (
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88,
89,
90,
91,
92,
93,
94,
95,
96,
97,
98,
99,
100,
101,
102,
103,
104,
105,
106,
107,
108,
109,
110,
111,
112,
113,
114,
115,
116,
117,
118,
119,
120,
121,
122,
123,
124,
125,
126,
127,
128,
129,
130,
131). ALCL may show B-cell, T-cell, or null-cell phenotype. T-cell and null-cell ALCL are discussed here.
Patients may present at any age, from infancy to adulthood. Either lymph node or skin involvement predominates. The bone marrow is affected in approximately 90% of cases at diagnosis. Rarely, the bone marrow is the only site of disease. Cases characterized by nodal involvement tend to have an aggressive course, with peripheral blood and bone marrow disease. Leukemia is especially common in the small cell variant of T-cell ALCL.
Laboratory studies may show anemia, neutropenia or neutrophilia, and absolute lymphocytosis. Neutrophilic leukemoid reaction has been reported.
Peripheral blood and bone marrow smears show abnormal lymphocytes which range from small, relatively nondescript tumor cells to large anaplastic cells with irregular to convoluted nuclei and prominent nucleoli. Either small cells or large cells may predominate. Smaller cells may show small, relatively bland nuclei and scanty cytoplasm. Larger cells show anaplastic features such as large, folded, lobated, bean-shaped, and embryo-shaped nuclei; wreath-shaped arrangements of multiple nuclei; and abundant vacuolated cytoplasm. A leukemic phase is particularly prominent in the small cell variant of ALCL. Abnormal lymphocytes may be difficult to identify among hematopoietic cells.
Histologic sections of the bone marrow show intrasinusoidal, interstitial, and’or diffuse lymphocytic infiltration. The abnormal lymphocytes are often admixed with inflammatory cells and fibrous tissue and may be difficult to identify among hematopoietic cells. Morphologic variants have been described, consisting predominantly of small cells, large nonanaplastic cells, and neoplastic cells admixed with numerous neutrophils, eosinophils, or histiocytes.
Other findings include myelodysplasia, apparently the result of abnormal cytokine secretion; and hemophagocytic syndrome, which may obscure the underlying malignancy.
Immunophenotyping usually shows a cytotoxic T-cell phenotype, with expression of CD2, CD3, CD5, CD7, CD30, CD45, S100 protein, ALK-1 (p80, NPM’ALK fusion protein), epithelial membrane antigen, granzyme B, perforin, and TIA-1. Compared to normal T-cells, expression of pan-T-cell antigens (CD2, CD3, CD5, CD7) may be reduced or absent. Either CD4 or CD8 may be expressed. CD11c, CD13, CD14, CD15, CD25, CD33, CD43, CD56, CD68, CD99, and epithelial membrane antigen may be expressed. ALK-1 may not be expressed. Antigen expression may vary between the peripheral blood and bone marrow. Cases expressing no recognizable T-cell or B-cell antigens are immunophenotypically null and diagnosed as anaplastic null-cell large cell lymphoma.
Genetic studies show, in most but not all cases, t(2;5)(p23;q35) involving ALK (anaplastic lymphoma kinase gene) at chromosome 2p23 and NPM at chromosome 5q35. Genetic variants involving ALK have been described. TCR and IgH gene rearrangements may also be present. Other genetic abnormalities have been reported.
The differential diagnosis includes drug reaction, anaplastic large B-cell lymphoma, Hodgkin lymphoma, acute leukemia, histiocytic neoplasms, and nonhematopoietic malignancies.