375e | Primary Immunodeficiencies Associated with (or Secondary to) Other Diseases |
There are an increasing number of conditions in which a primary immunodeficiency (PID) has been described as one facet of a more complex disease setting. It is essential to consider associated diseases when a PID is identified as the primary manifestation and, conversely, not neglect the potentially harmful consequences of a PID that could be masked by other manifestations of a particular syndrome.
Below is a short description of these syndromes in which the PID is classified according to the arm of the immune system that is affected.
1. Primary Immunodeficiencies of the Innate Immune System
a) Several severe congenital neutropenia (SCN) syndromes can be associated with malformations. The recently described SCN disease caused by glucose-6-phosphatase deficiency (G6PC3) can be associated with heart and urogenital malformations. The related glycogenesis Ib disease combines SCN with hypoglycemia and hepatosplenomegaly. Some HAX-1 gene mutations lead to neurocognitive impairments as well as SCN. Barth syndrome combines SCN with cardiomyopathy. Lastly, Shwachman syndrome is a known autosomal recessive entity (caused by mutation of the SBDS gene) in which the defect in granulopoiesis can extend to the other hematopoietic lineages; short stature, bone metaphyseal dysplasia, and exocrine pancreatic insufficiency are known hallmarks of this condition.
b) Syndromic asplenia combines the risk of infection with heart defects and situs inversus.
c) Leukocyte adhesion deficiency (LAD) type II includes growth retardation and impairment of cognitive development.
d) A few patients with X-linked chronic granulomatous disease present with a contiguous gene deletion syndrome that can include the McLeod phenotype, which is characterized by anemia, acanthocytosis, and a severe risk of immune reaction against donor red cells because the patient’s red cells do not express the Kell antigen. The McLeod phenotype also can result in a neurologic disease.
e) X-linked nuclear factor-κB (NF-κB) essential modulator (NEMO) deficiency provokes not only a variable set of deficiencies of both innate and adaptive immunity but also mild osteopetrosis, lymphedema, and, more frequently, anhydrotic ectodermal dysplasia, dysmorphic facies, and abnormal conical teeth. The last finding is often helpful in the diagnosis of that condition.
2. Primary Immunodeficiencies of the Adaptive Immune System
a) T cell primary immunodeficiencies. Reticular dysgenesis, a rare severe combined immunodeficiency (SCID) characterized by T lymphopenia and agranulocytosis, can cause sensorineural deafness. Coronin A deficiency is another SCID variant that can be associated with behavioral disorders because the Coronin A gene is located in a genome area known to have been deleted in some patients with this disorder. The lack of enzymes of purine metabolism (adenosine deaminase and purine nucleoside phosphorylase) provokes not only profound T cell lymphocytopenia but also neurologic impairment, including dysautonomia and abnormalities of cognitive development of variable intensity, in many patients. The neurologic impairment can persist after hematopoietic stem cell transplantation (HSCT). Mild chondrodysplasia is a common finding in adenosine deaminase (ADA) deficiency and, indeed, can help the physician arrive at a final diagnosis.
b) Primary thymic defects. DiGeorge syndrome is a complex embryopathy that is caused by hemizygous interstitial deletion of chromosome 22, leading to multiple developmental defects, including conotruncal defects, hypoparathyroidism, and dysmorphic syndrome. Although a profound T cell immunodeficiency is rare in DiGeorge syndrome (∼1% of cases), failure to recognize this feature is likely to have a fatal outcome. Similarly, some forms of the related CHARGE syndrome (mutation of the CHD7 gene) also cause a profound T cell immunodeficiency.
c) T cell primary immunodeficiencies related to calcium influx defects. Recently, rare T cell PIDs were found to be caused by defective store-operated entry of calcium ions into T and B lymphocytes after antigen stimulation. These defects (caused by ORA-1 and STIM-1 deficiencies) also lead to anhydrotic ectodermal dysplasia, abnormal teeth, and, above all, a nonprogressive muscle disease characterized by excessive fatigue.
d) DNA repair defects. Several genetic defects impair DNA repair pathways. Many lead to combined T and B lymphocyte PIDs in a syndromal setting of varying complexity. The most common is ataxia-telangiectasia (AT), an autosomal recessive disorder with an incidence of 1 in 40,000 live births; AT causes a B cell immunodeficiency (low IgA, IgG2 deficiency, and low antibody production) that often requires immunoglobulin replacement therapy.
AT is associated with a progressive T cell immunodeficiency. As the condition’s name suggests, the hallmark features are telangiectasia and cerebellar ataxia.
These manifestations may not be detectable before age 3–4 years, and so AT should be considered in young children with IgA deficiency and problematic infections. Diagnosis is based on a cytogenetic analysis showing excessive chromosomal rearrangements (mostly affecting chromosomes 7 and 14) in lymphocytes. AT is caused by mutation of the gene encoding the ATM protein, a kinase that plays a major role in the detection of DNA lesions and the organization of DNA repair (or cell death if the lesions are too numerous) by triggering several different pathways. Overall, AT is a progressive disease that carries a very high risk of lymphoma, leukemia, and (during adulthood) carcinomas. A variant of AT (AT-like disease) is caused by mutation of the MRE11 gene.
Nijmegen breakage syndrome (NBS) is a less common condition that also results from chromosome instability (and the same cytogenetic abnormalities as in AT). It is characterized by a severe T and B cell combined immunodeficiency with autosomal recessive inheritance. Subjects with NBS exhibit microcephaly and a birdlike face but neither ataxia nor telangiectasia. The risk of malignancies is also very high. Nijmegen breakage syndrome results from a deficiency in Nibrin (NBS1, a protein associated with MRE11 and Rad50 that is involved in checking DNA lesions) caused by hypomorphic mutations.
Severe forms of dyskeratosis congenita (also known as Hoyeraal-Hreidersson syndrome) combine a progressive immunodeficiency that can include an absence of B and natural killer cell (NK) lymphocytes, progressive bone marrow failure, microcephaly, in utero growth retardation, and gut disease. The disease can be X-linked or, more rarely, autosomal recessive. It is caused by the mutation of genes encoding telomere maintenance proteins, including dyskerin (DKC1).
Bloom syndrome (helicase deficiency) combines a typical dysmorphic syndrome with growth retardation, skin lesions, and a mild immunodeficiency that also can be found in some patients with Fanconi’s anemia.
Rare forms of combined T and B cell immunodeficiencies with autosomal recessive inheritance are associated in more complex syndromes with microcephaly, failure to grow, and a variable dysmorphic syndrome. These disorders are caused by mutation of the genes that encode DNA ligase 4 and Cernunnos (XLF), both of which are members of the nonhomologous end-joining DNA repair pathway.
The Vici syndrome combines callosal agenesis, cataracts, cardiomyopathy, hyperpigmentation, and a combined immunodeficiency. It is caused by biallelic EPG5 gene mutation that results in defective autophagy.
Lastly, immunodeficiency, centromere instability, and facial anomalies (ICF) syndrome is a complex autosomal recessive syndrome that variably combines a mild T cell immunodeficiency and a more severe B cell immunodeficiency with a coarse face, intestinal disease, and mild mental retardation. A cytogenetic diagnostic feature is the presence of multiradial chromosomes (most frequently chromosomes 1, 9, and 16) caused by DNA defective methylation. The syndrome is a result of either DNA methyltransferase DNMT3B deficiency or ZBTB24 deficiency.
e) Growth hormone insensitivity syndrome (Laron dwarfism) with combined primary immunodeficiency. Mutations in the STAT5b gene, which encodes a transcription factor involved in signaling downstream of the growth hormone receptor and the interleukin 2 (IL-2) receptor, lead to susceptibility to infection because of a partial, functional T cell immunodeficiency associated with autoimmune manifestations. The autoimmune manifestations probably result from defective generation/activation of regulatory T cells.
f) Hyper-IgE syndrome (autosomal dominant form). Hyper-IgE syndrome is a complex disorder that combines skin infections, inflammation, and susceptibility to bacterial and fungal infections of skin and lungs, often with pneumatoceles, with characteristic syndromic signs such as facial dysmorphy, defective loss of primary teeth, hyperextensibility, scoliosis, and osteoporosis. Elevated serum IgE levels are typical of hyper-IgE syndrome. The recently reported defects in TH17 effector responses account, at least in part, for the vulnerability to specific infections. This condition is caused by heterozygous (dominant) mutation of the gene encoding the transcription factor STAT3, which is required in a number of signaling pathways downstream of cytokine/cytokine receptor interactions (notably for IL-6 and IL21).
g) Primary immunodeficiencies with bone disease. The autosomal recessive cartilage hair hypoplasia (CHH) disease is characterized by short-limb dwarfism, metaphyseal dysostosis, and sparse hair, together with a combined T and B cell PID of variable intensity, ranging from quasi-SCID to an absence of clinically significant immunodeficiency. The condition can predispose to erythroblastopenia, autoimmunity, and tumors. It is caused by mutations in the RMRP gene for a noncoding ribosome-associated RNA.
Schimke immunoosseous dysplasia is a rare autosomal recessive condition characterized by severe T and B cell immunodeficiency with spondyloepiphyseal dysplasia, growth retardation, and kidney and vascular diseases. It is the consequence of mutations in the SMARCAL1 gene. The function of the gene product may be related to DNA repair.
h) Venoocclusive disease with immunodeficiency (VODI syndrome) is a rare autosomal recessive condition predominantly found in populations originating from Lebanon. It combines severe hepatic venoocclusive disease with usually mild T cell immunodeficiency and panhypogammaglobulinemia. It is caused by a deficiency in a nuclear protein, Sp110.
3. B Cell Primary Immunodeficiencies Hypogammaglobulinemia can be associated with chromosomal defects such as trisomy 18 and Jacobsen syndrome (hemizygous deletion of part of the long arm of chromosome 11). A rare biallelic deficiency of the mismatch repair protein PMS2 leads to a partial deficiency in Ig class switch recombination in patients at a very high risk of cancer in general and colon carcinomas and lymphomas in particular. Transcobalamin deficiency disturbs vitamin B12 transport and therefore impairs hematopoiesis. Hypogammaglobulinemia is easily corrected by vitamin B12 administration and can be a characteristic of this very rare disorder.
4. Primary Immunodeficiencies Affecting Regulatory Pathways Several inherited disorders that lead to hemophagocytic lymphohistiocytosis (HLH) also have features that are important in terms of both diagnosis and prognosis. Three of these disorders—Griscelli syndrome, Chédiak-Higashi syndrome, and the Hermansky-Pudlak type II syndrome—are characterized by partial albinism and silvery hair appearance that can facilitate diagnosis. Hermansky-Pudlak type II also can be a bleeding disorder if platelet aggregation is defective. Chédiak-Higashi syndrome also is characterized by an early-onset progressive neurologic disorder with impaired cognitive development and motor and sensory deficiencies, culminating in a generalized encephalopathy. The encephalopathy is not prevented or arrested by allogeneic HSCT even when the HLH risk is controlled.
5. Primary Immunodeficiencies Associated with Other Conditions Predisposition to infection, notably severe disseminated opportunistic infections including nontuberculous mycobacterial infections, can be associated with autoantibodies against interferon γ as observed in Asia.
A number of conditions can cause PIDs indirectly. For example, hypercatabolism in patients with Steinert’s disease may cause hypogammaglobulinemia. Intestinal lymphangiectasia that includes both immunoglobulin and naive T cell loss and can expose the patient to a significant infectious risk. Urinary IgG loss may result from severe nephritic syndromes.
A number of drugs, including antimalarials, captopril, penicillamine, phenytoin, and sulfasalazine, can induce predominantly IgA hypogammaglobulinemia in (probably predisposed) adults.
One also should also consider (1) diseases that are not thought to be PIDs but include the occurrence of recurrent infections and (2) genetic defects of the immune system that lead to other clinical manifestations. A very good example of the first group is cystic fibrosis (CF). Despite having a functionally normal immune system, patients with CF develop protracted bacterial respiratory tract infections, notably Pseudomonas aeruginosa colonization. This bacterium can incapacitate innate immune responses and cause unremitting inflammation that further facilitates infection. An example of the second group is primary alveolar proteinosis, which is caused by a defect in surfactant clearance by alveolar macrophages. The condition results from mutation of the gene encoding the granulocyte-macrophage colony-stimulating factor receptor α.
SECTION 2 | DISORDERS OF IMMUNE-MEDIATED INJURY |
376 | Allergies, Anaphylaxis, and Systemic Mastocytosis |
The term atopy implies a tendency to manifest asthma, rhinitis, urticaria, and atopic dermatitis alone or in combination, in association with the presence of allergen-specific IgE. However, individuals without an atopic background may also develop hypersensitivity reactions, particularly urticaria and anaphylaxis, associated with the presence of IgE. Inasmuch as the mast cell is a key effector cell in allergic rhinitis and asthma, and the dominant effector in urticaria, anaphylaxis, and systemic mastocytosis, its developmental biology, activation pathway, product profile, and target tissues will be considered in the introduction to these clinical disorders.
The binding of IgE to human mast cells and basophils, a process termed sensitization, prepares these cells for subsequent antigen-specific activation. The high-affinity Fc receptor for IgE, designated FcεRI, is composed of one α, one β, and two disulfide-linked γ chains, which together cross the plasma membrane seven times. The α chain is responsible for IgE binding, and the β and γ chains provide for signal transduction that follows the aggregation of the sensitized tetrameric receptors by polymeric antigen. The binding of IgE stabilizes the α chain at the plasma membrane, thus increasing the density of FcεRI receptors at the cell surface while sensitizing the cell for effector responses. This accounts for the correlation between serum IgE levels and the numbers of FcεRI receptors detected on circulating basophils.
Signal transduction is initiated through the action of a Src family–related tyrosine kinase termed Lyn that is constitutively associated with the β chain. Lyn transphosphorylates the canonical immunoreceptor tyrosine-based activation motifs (ITAMs) of the β and γ chains of the receptor, resulting in recruitment of more active Lyn to the β chain and of Syk tyrosine kinase. The phosphorylated tyrosines in the ITAMs function as binding sites for the tandem src homology two (SH2) domains within Syk. Syk activates not only phospholipase Cγ, which associates with the linker of activated T cells at the plasma membrane, but also phosphatidylinositol 3-kinase to provide phosphatidylinositol-3,4,5-trisphosphate, which allows membrane targeting of the Tec family kinase Btk and its activation by Lyn. In addition, the Src family tyrosine kinase Fyn becomes activated after aggregation of IgE receptors and phosphorylates the adapter protein Gab2 that enhances activation of phosphatidylinositol 3-kinase. Indeed, this additional input is essential for mast cell activation, but it can be partially inhibited by Lyn, indicating that the extent of mast cell activation is in part regulated by the interplay between these Src family kinases. Activated phospholipase Cγ cleaves phospholipid membrane substrates to provide inositol-1,4,5-trisphosphate (IP3) and 1,2-diacylglycerols (1,2-DAGs) so as to mobilize intracellular calcium and activate protein kinase C, respectively. The subsequent opening of calcium-regulated activated channels provides the sustained elevations of intracellular calcium required to recruit the mitogen-activated protein kinases, ERK, JNK, and p38 (serine/threonine kinases), which provide cascades to augment arachidonic acid release and to mediate nuclear translocation of transcription factors for various cytokines. The calcium ion–dependent activation of phospholipases cleaves membrane phospholipids to generate lysophospholipids, which, like 1,2-DAG, may facilitate the fusion of the secretory granule perigranular membrane with the cell membrane, a step that releases the membrane-free granules containing the preformed mediators of mast cell effects.
The secretory granule of the human mast cell has a crystalline structure, unlike mast cells of lower species. IgE-dependent cell activation results in solubilization and swelling of the granule contents within the first minute of receptor perturbation; this reaction is followed by the ordering of intermediate filaments about the swollen granule, movement of the granule toward the cell surface, and fusion of the perigranular membrane with that of other granules and with the plasmalemma to form extracellular channels for mediator release while maintaining cell viability.
In addition to exocytosis, aggregation of FcεRI initiates two other pathways for generation of bioactive products, namely, lipid mediators and cytokines. The biochemical steps involved in expression of such cytokines as tumor necrosis factor α (TNF-α), interleukin (IL) 1, IL-6, IL-4, IL-5, IL-13, granulocyte-macrophage colony-stimulating factor (GM-CSF), and others, including an array of chemokines, have not been specifically defined for mast cells. Inhibition studies of cytokine production (IL-1β, TNF-α, and IL-6) in mouse mast cells with cyclosporine or FK506 reveal binding to the ligand-specific immunophilin and attenuation of the calcium ion- and calmodulin-dependent serine/threonine phosphatase, calcineurin.
Lipid mediator generation (Fig. 376-1) involves translocation of calcium ion–dependent cytosolic phospholipase A2 to the outer nuclear membrane, with subsequent release of arachidonic acid for metabolic processing by the distinct prostanoid and leukotriene pathways. The constitutive prostaglandin endoperoxide synthase-1 (PGHS-1/cyclooxygenase-1) and the de novo inducible PGHS-2 (cyclooxygenase-2) convert released arachidonic acid to the sequential intermediates, prostaglandins G2 and H2. The glutathione-dependent hematopoietic prostaglandin D2 (PGD2) synthase then converts PGH2 to PGD2, the predominant mast cell prostanoid. The PGD2 receptor DP1 is expressed by platelets and epithelial cells, whereas DP2 is expressed by TH2 lymphocytes, eosinophils, and basophils. Mast cells also generate thromboxane A2 (TXA2), a short lived but powerful mediator that induces bronchoconstriction and platelet activation through the T prostanoid (TP) receptor.
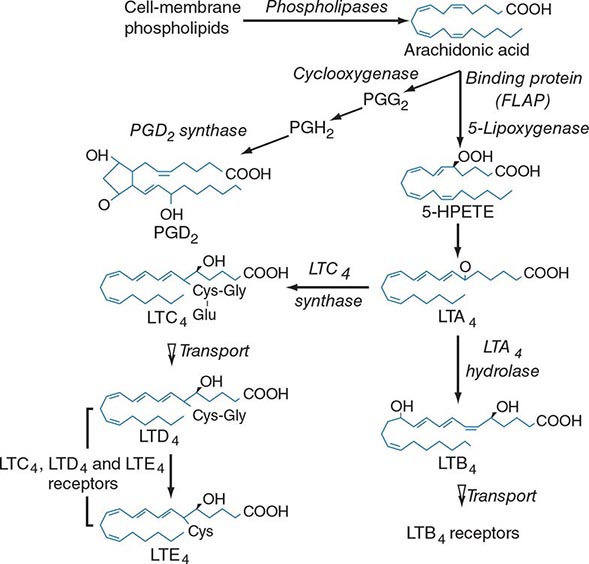
FIGURE 376-1 Pathways for biosynthesis and release of membrane-derived lipid mediators from mast cells. In the 5-lipoxygenase pathway, leukotriene A4 (LTA4) is the intermediate from which the terminal-pathway enzymes generate the distinct final products, leukotriene C4 (LTC4) and leukotriene B4 (LTB4), which leave the cell by separate saturable transport systems. Gamma glutamyl transpeptidase and a dipeptidase then cleave glutamic acid and glycine from LTC4 to form LTD4 and LTE4, respectively. The major mast cell product of the cyclooxygenase system is PGD2.
For leukotriene biosynthesis, the released arachidonic acid is metabolized by 5-lipoxygenase (5-LO) in the presence of an integral nuclear membrane protein, 5-LO activating protein (FLAP). The calcium ion–dependent translocation of 5-LO to the nuclear membrane converts the arachidonic acid to the sequential intermediates, 5-hydroperoxyeicosatetraenoic acid (5-HPETE) and leukotriene (LT) A4. LTA4 is conjugated with reduced glutathione by LTC4 synthase, an integral nuclear membrane protein homologous to FLAP. Intracellular LTC4 is released by a carrier-specific export step for extracellular metabolism to the additional cysteinyl leukotrienes, LTD4 and LTE4, by the sequential removal of glutamic acid and glycine. Alternatively, cytosolic LTA4 hydrolase converts some LTA4 to the dihydroxy leukotriene LTB4, which also undergoes specific export. Two receptors for LTB4, BLT1 and BLT2, mediate chemotaxis of human neutrophils. Two receptors for the cysteinyl leukotrienes, CysLT1 and CysLT2, are present on smooth muscle of the airways and the microvasculature and on hematopoietic cells such as macrophages, eosinophils, and mast cells. Whereas the CysLT1 receptor has a preference for LTD4 and is blocked by the receptor antagonists in clinical use, the CysLT2 receptor is equally responsive to LTD4 and LTC4, is unaffected by these antagonists, and is a negative regulator of the function of the CysLT1 receptor. LTD4, acting at CysLT1 receptors, is the most potent known bronchoconstrictor, whereas LTE4 induces a vascular leak and mediates the recruitment of eosinophils to the bronchial mucosa. Studies in gene-deleted mice indicate the existence of additional receptors for LTE4. The lysophospholipid formed during the release of arachidonic acid from 1-O-alkyl-2-acyl-sn-glyceryl-3-phosphorylcholine can be acetylated in the second position to form platelet-activating factor (PAF). Serum levels of PAF correlated positively with the severity of anaphylaxis to peanut in a recent study, whereas the levels of PAF acetyl hydrolase (a PAF-degrading enzyme) were inversely related to the same outcome.
Unlike most other cells of bone marrow origin, mast cells circulate as committed progenitors lacking their characteristic secretory granules. These committed progenitors express c-kit, the receptor for stem cell factor (SCF). Unlike most other lineages, they retain and increase c-kit expression with maturation. The SCF interaction with c-kit is an absolute requirement for the development of constitutive tissue mast cells residing in skin and connective tissue sites and for the accumulation of mast cells at mucosal surfaces during TH2-type immune responses. Several T cell-derived cytokines (IL-3, IL-4, IL-5, and IL-9) can potentiate SCF-dependent mast cell proliferation and/or survival in vitro in mice and humans. Indeed, mast cells are absent from the intestinal mucosa in clinical T cell deficiencies, but are present in the submucosa. Based on the immunodetection of secretory granule neutral proteases, mast cells in the lung parenchyma and intestinal mucosa selectively express tryptase, and those in the intestinal and airway submucosa, perivascular spaces, skin, lymph nodes, and breast parenchyma express tryptase, chymase, and carboxypeptidase A (CPA). In the mucosal epithelium of severe asthmatics, mast cells can express tryptase and CPA without chymase. The secretory granules of mast cells selectively positive for tryptase exhibit closed scrolls with a periodicity suggestive of a crystalline structure by electron microscopy, whereas the secretory granules of mast cells with multiple proteases are scroll-poor, with an amorphous or lattice-like appearance.
Mast cells are distributed at cutaneous and mucosal surfaces and in submucosal tissues about venules and could influence the entry of foreign substances by their rapid response capability (Fig. 376-2). Upon stimulus-specific activation and secretory granule exocytosis, histamine and acid hydrolases are solubilized, whereas the neutral proteases, which are cationic, remain largely bound to the anionic proteoglycans, heparin and chondroitin sulfate E, with which they function as a complex. Histamine and the various lipid mediators (PGD2, LTC4/D4/E4, PAF) alter venular permeability, thereby allowing influx of plasma proteins such as complement and immunoglobulins, whereas LTB4 mediates leukocyte–endothelial cell adhesion and subsequent directed migration (chemotaxis). The accumulation of leukocytes and plasma opsonins facilitates defense of the microenvironment. The inflammatory response can also be detrimental, as in asthma, where the smooth-muscle constrictor activity of the cysteinyl leukotrienes is evident and much more potent than that of histamine.
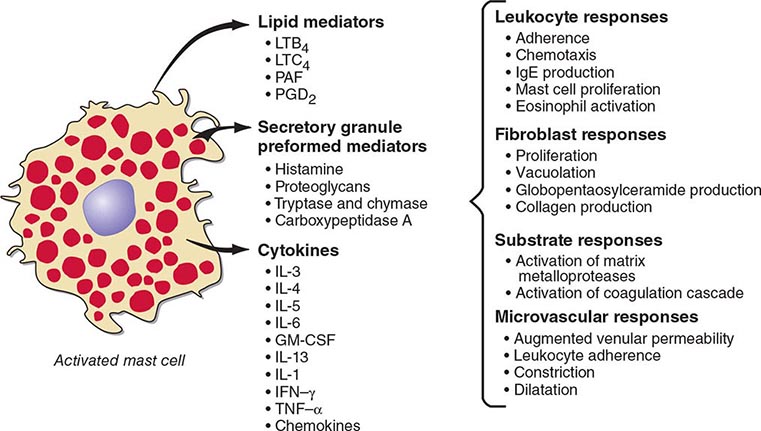
FIGURE 376-2 Bioactive mediators of three categories generated by IgE-dependent activation of murine mast cells can elicit common but sequential target cell effects leading to acute and sustained inflammatory responses. GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; IFN, interferon; LT, leukotriene; PAF, platelet-activating factor; PGD2, prostaglandin D2; TNF, tumor necrosis factor.
The cellular component of the mast cell–mediated inflammatory response is augmented and sustained by cytokines and chemokines. IgE-dependent activation of human skin mast cells in situ elicits TNF-α production and release, which in turn induces endothelial cell responses favoring leukocyte adhesion. Similarly, activation of purified human lung mast cells or cord blood–derived cultured mast cells in vitro results in substantial production of proinflammatory (TNF-α) and immunomodulatory cytokines (IL-4, IL-5, IL-13) and chemokines. Bronchial biopsy specimens from patients with asthma reveal that mast cells are immunohistochemically positive for IL-4 and IL-5, but that the predominant localization of IL-4, IL-5, and GM-CSF is to T cells, defined as TH2 by this profile. IL-4 modulates the T cell phenotype to the TH2 subtype, determines the isotype switch to IgE (as does IL-13), and upregulates FcεRI-mediated expression of cytokines by mast cells based on in vitro studies.
An immediate and late cellular phase of allergic inflammation can be induced in the skin, nose, or lung of some allergic humans with local allergen challenge. The immediate phase in the nose involves pruritus and watery discharge; in the lung, it involves bronchospasm and mucus secretion; and in the skin, it involves a wheal-and-flare response with pruritus. The reduced nasal patency, reduced pulmonary function, or erythema with swelling at the skin site in a late-phase response at 6–8 h is associated with biopsy findings of infiltrating and activated TH2 cells, eosinophils, basophils, and some neutrophils. The progression from early mast cell activation to late cellular infiltration has been used as an experimental surrogate of rhinitis or asthma. However, in asthma, there is an intrinsic hyperreactivity of the airways independent of the associated inflammation. Moreover, early- and late-phase responses (at least in the lung) are far more sensitive to blockade of IgE-dependent mast cell activation (or actions of histamine and cysteinyl leukotrienes) than are spontaneous or virally induced asthma exacerbations.
Consideration of the mechanism of immediate-type hypersensitivity diseases in the human has focused largely on the IgE-dependent recognition of otherwise innocuous substances. A region of chromosome 5 (5q23-31) contains genes implicated in the control of IgE levels including IL-4 and IL-13, as well as IL-3 and IL-9, which are involved in mucosal mast cell hyperplasia, and IL-5 and GM-CSF, which are central to eosinophil development and their enhanced tissue viability. Genes with linkage to the specific IgE response to particular allergens include those encoding the major histocompatibility complex (MHC) and certain chains of the T cell receptor (TCR-αδ). The complexity of atopy and the associated diseases includes susceptibility, severity, and therapeutic responses, each of which is among the separate variables modulated by both innate and adaptive immune stimuli.
The induction of allergic disease requires sensitization of a predisposed individual to a specific allergen. The greatest propensity for the development of atopic allergy occurs in childhood and early adolescence. The allergen is processed by antigen-presenting cells of the monocytic lineage (particularly dendritic cells) located throughout the body at surfaces that contact the outside environment, such as the nose, lungs, eyes, skin, and intestine. These antigen-presenting cells present the epitope-bearing peptides via their MHC to T helper cells and their subsets. The T cell response depends both on cognate recognition and on the cytokine microenvironment provided by the antigen-presenting dendritic cells, with IL-4 directing a TH2 subset, interferon (IFN) γ a TH1 profile, and IL-6 with transforming growth factor β (TGF-β) a TH17 subset. Allergens not only present antigenic epitopes via dendritic cells but also contain pattern recognition ligands that facilitate the immune response by direct initiation of cytokine generation from innate cell types such as basophils, mast cells, eosinophils, and others. The TH2 response is associated with activation of specific B cells that can also present allergens or that transform into plasma cells for antibody production. Synthesis and release into the plasma of allergen-specific IgE results in sensitization of FcεR1-bearing cells such as mast cells and basophils, which become activated on exposure to the specific allergen. In certain diseases, including those associated with atopy, the monocyte and eosinophil populations can express a trimeric FcεR1, which lacks the β chain, and yet respond to its aggregation. An additional recently recognized class of c-kit-expressing innate cells (termed nuocytes, natural helper cells, or group 2 innate lymphoid cells) can generate large quantities of IL-5 and IL-13 during antihelminth responses, are prominent in nasal polyps from humans, and could well contribute to inflammation in allergic diseases.
ANAPHYLAXIS
DEFINITION
Life-threatening anaphylactic responses of sensitized humans occur within minutes after systemic exposure to specific antigen. They are manifested by respiratory distress due to laryngeal edema and/or intense bronchospasm, often followed by vascular collapse, or by shock without antecedent respiratory difficulty. Cutaneous manifestations exemplified by pruritus and urticaria with or without angioedema are characteristic of such systemic anaphylactic reactions. Gastrointestinal manifestations include nausea, vomiting, crampy abdominal pain, and diarrhea.
PREDISPOSING FACTORS AND ETIOLOGY
There is no convincing evidence that age, sex, race, or geographic location predisposes a human to anaphylaxis except through exposure to specific immunogens. According to most studies, atopy does not predispose individuals to anaphylaxis from penicillin therapy or venom of a stinging insect but is a risk factor for allergens in food or latex. Risk factors for a poor outcome, however, include older age, use of beta blockers, and the presence of preexisting asthma. Severe hymenoptera anaphylaxis (generally with prominent hypotension) can be a presenting feature of underlying systemic mastocytosis. Additionally, some individuals suffering from recurrent episodes of idiopathic anaphylaxis possess morphologically aberrant mast cells in their bone marrow that express a mutant, constitutively active form of c-kit, even without evidence of frank mastocytosis.
The materials capable of eliciting the systemic anaphylactic reaction in humans include the following: heterologous proteins in the form of hormones (insulin, vasopressin, parathormone); enzymes (trypsin, chymotrypsin, penicillinase, streptokinase); pollen extracts (ragweed, grass, trees); nonpollen allergen extracts (dust mites, dander of cats, dogs, horses, and laboratory animals); food (peanuts, milk, eggs, seafood, nuts, grains, beans, gelatin in capsules); monoclonal antibodies; occupation-related products (latex rubber products); Hymenoptera venom (yellow jacket, yellow and white-faced hornets, paper wasp, honey bee, imported fire ants); polysaccharides such as dextran and thiomersal as a vaccine preservative; drugs such as protamine; antibiotics (penicillins, cephalosporins, amphotericin B, nitrofurantoin, quinolones); chemotherapy agents (carboplatin, paclitaxel, doxorubicin); local anesthetics (procaine, lidocaine); muscle relaxants (suxamethonium, gallamine, pancuronium); vitamins (thiamine, folic acid); diagnostic agents (sodium dehydrocholate, sulfobromophthalein); biologics (omalizumab, rituximab, etanercept); and occupation-related chemicals (ethylene oxide). Drugs function as haptens that form immunogenic conjugates with host proteins. The conjugating hapten may be the parent compound, a nonenzymatically derived storage product, or a metabolite formed in the host. Recombinant biologics can also induce the formation of IgE against the proteins or against glycosylated structures that serve as immunogens. Most recently, outbreaks of anaphylaxis to the anti-epidermal growth factor antibody cetuximab were reported in association with elevated titers of serum IgE to alpha-1,3-galactose, an oligosaccharide found on certain nonprimate proteins. Alpha-galactose antibodies also account for some episodes of delayed anaphylaxis to beef, lamb, and pork.
PATHOPHYSIOLOGY AND MANIFESTATIONS
Individuals differ in the time of appearance of symptoms and signs, but the hallmark of the anaphylactic reaction is the onset of some manifestation within seconds to minutes after introduction of the antigen (with the exception of alpha-galactose allergy), generally by injection or less commonly by ingestion. There may be upper or lower airway obstruction or both. Laryngeal edema may be experienced as a “lump” in the throat, hoarseness, or stridor, whereas bronchial obstruction is associated with a feeling of tightness in the chest and/or audible wheezing. Patients with asthma are predisposed to severe involvement of the lower airways and increased mortality. Flushing with diffuse erythema and a feeling of warmth may occur. A characteristic feature is the eruption of well-circumscribed, discrete cutaneous wheals with erythematous, raised, serpiginous borders and blanched centers. These urticarial eruptions are intensely pruritic and may be localized or disseminated. They may coalesce to form giant hives, and they seldom persist beyond 48 h. A localized, nonpitting, deeper edematous cutaneous process, angioedema, may also be present. It may be asymptomatic or cause a burning or stinging sensation. Angioedema of the bowel wall may cause sufficient intravascular volume depletion to precipitate cardiovascular collapse.
In fatal cases with clinical bronchial obstruction, the lungs show marked hyperinflation on gross and microscopic examination. The microscopic findings in the bronchi, however, are limited to luminal secretions, peribronchial congestion, submucosal edema, and eosinophilic infiltration, and the acute emphysema is attributed to intractable bronchospasm that subsides with death. The angioedema resulting in death by mechanical obstruction occurs in the epiglottis and larynx, but the process also is evident in the hypopharynx and to some extent in the trachea. On microscopic examination, there is wide separation of the collagen fibers and the glandular elements; vascular congestion and eosinophilic infiltration also are present. Patients dying of vascular collapse without antecedent hypoxia from respiratory insufficiency have visceral congestion with a presumptive loss of intravascular fluid volume. The associated electrocardiographic abnormalities, with or without infarction, in some patients may reflect a primary cardiac event mediated by mast cells (which are prominent near the coronary vessels) or may be secondary to a critical reduction in blood volume.
The angioedematous and urticarial manifestations of anaphylaxis have been attributed to the release of endogenous histamine. A role for the cysteinyl leukotrienes in causing marked bronchiolar constriction seems likely. Vascular collapse without respiratory distress in response to experimental challenge with the sting of a hymenopteran was associated with marked and prolonged elevations in blood histamine and intravascular coagulation and kinin generation. The finding that patients with systemic mastocytosis and episodic vascular collapse excrete large amounts of PGD2 metabolites in addition to histamine suggests that PGD2 is also of importance in the hypotensive anaphylactic reactions. As noted, serum PAF levels correlate with severity of anaphylaxis and are inversely proportional to the constitutive level of the acetylhydrolase involved in PAF inactivation. The actions of the array of mast cell–derived mediators are likely additive or synergistic at the target tissues.
DIAGNOSIS
The diagnosis of an anaphylactic reaction depends on a history revealing the onset of symptoms and signs within minutes after the responsible material is encountered. It is appropriate to rule out a complement-mediated immune complex reaction, an idiosyncratic response to a nonsteroidal anti-inflammatory drug (NSAID), or the direct effect of certain drugs or diagnostic agents on mast cells. Intravenous administration of a chemical mast cell–degranulating agent, including opiate derivatives and radiographic contrast media, may elicit generalized urticaria, angioedema, and a sensation of retrosternal oppression with or without clinically detectable bronchoconstriction or hypotension. In the transfusion anaphylactic reaction that occurs in patients with IgA deficiency, the responsible specificity resides in IgG or IgE anti-IgA; the mechanism of the reaction mediated by IgG anti-IgA is presumed to be complement activation with secondary mast cell participation.
The presence of specific IgE in the blood of patients with systemic anaphylaxis was demonstrated historically by passive transfer of the serum intradermally into a normal recipient, followed 24 h later by antigen challenge into the same site, with subsequent development of a wheal and flare (the Prausnitz-Küstner reaction). In current clinical practice, immunoassays using purified or recombinant antigens can demonstrate the presence of specific IgE in the serum of patients with anaphylactic reactions, and skin testing may be performed after the patient has recovered to elicit a local wheal and flare in response to the putative antigen. Elevations of tryptase levels in serum implicate mast cell activation in a systemic reaction and are particularly informative for anaphylaxis with episodes of hypotension during general anesthesia or when there has been a fatal outcome. However, because of the short half-life of tryptase, elevated levels are best detected within 4 h of a systemic reaction. Moreover, anaphylactic reactions to foods characteristically are not associated with elevations in serum tryptase.
PREVENTION
Prevention of anaphylaxis must take into account the sensitivity of the individual, the dose and character of the diagnostic or therapeutic agent, and the effect of the route of administration on the rate of absorption. Beta blockers are relatively contraindicated in persons at risk for anaphylactic reactions, especially those sensitive to Hymenoptera venom or those undergoing immunotherapy for respiratory system allergy. If there is a definite history of a past anaphylactic reaction to a medication, it is advisable to select a structurally unrelated agent. A knowledge of cross-reactivity among agents is critical since, for example, cephalosporins have a cross-reactive ring structure with the penicillins. When skin testing, a prick or scratch skin test should precede an intradermal test, since the latter has a higher risk of causing anaphylaxis. These tests should be performed before the administration of certain materials that are likely to elicit anaphylactic reactions, such as allergenic extracts. Skin testing for antibiotics or chemotherapeutic agents should be performed only on patients with a positive clinical history consistent with an IgE-mediated reaction and in imminent need of the antibiotic in question; skin testing is of no value for non-IgE-mediated eruptions. With regard to penicillin, two-thirds of patients with a positive reaction history and positive skin tests to benzylpenicilloyl-polylysine (BPL) and/or the minor determinant mixture (MDM) of benzylpenicillin products experience allergic reactions with treatment, and these reactions are almost uniformly of the anaphylactic type in those patients with minor determinant reactivity. Even patients without a history of previous clinical reactions have a 2–6% incidence of positive skin tests to the two test materials, and about 3 per 1000 with a negative history experience anaphylaxis with therapy, with a mortality of about 1 per 100,000.
If an agent carrying a risk of eliciting an anaphylactic response is required because a non-cross-reactive alternative is not available, desensitization can be performed with most antibiotics and other classes of therapeutic agents by the IV, SC, or PO route. Typically, graded quantities of the drug are given by the selected route starting below the threshold dose for an adverse reaction and then doubling each dose until a therapeutic dosage is achieved. Due to the risk of systemic anaphylaxis during the course of desensitization, such a procedure should be performed under the supervision of a specialist and in a setting in which resuscitation equipment is at hand and an IV line is in place. Once a desensitized state is achieved, it is critical to continue administration of the therapeutic agent at regular intervals throughout the treatment period to prevent the reestablishment of a significant pool of sensitized cells.
A different form of protection involves the development of blocking antibody of the IgG class, which protects against Hymenoptera venom–induced anaphylaxis by interacting with antigen so that less reaches the sensitized tissue mast cells. The maximal risk for systemic anaphylactic reactions in persons with Hymenoptera sensitivity occurs in association with a currently positive skin test. Although there is little cross-reactivity between honey bee and yellow jacket venoms, there is a high degree of cross-reactivity between yellow jacket venom and the rest of the vespid venoms (yellow or white-faced hornets and wasps). Prevention involves modification of outdoor activities to exclude bare feet, wearing perfumed toiletries, eating in areas attractive to insects, clipping hedges or grass, and hauling away trash or fallen fruit. As with each anaphylactic sensitivity, the individual should wear an informational bracelet and have immediate access to an unexpired autoinjectable epinephrine kit. Venom immunotherapy for 5 years can induce a state of resistance to sting reactions that is independent of serum levels of specific IgG or IgE. For children under the age of 10 with a systemic reaction limited to skin, the likelihood of progression to more serious respiratory or vascular manifestations is low, and thus immunotherapy is not recommended.
URTICARIA AND ANGIOEDEMA
DEFINITION
Urticaria and angioedema may appear separately or together as cutaneous manifestations of localized nonpitting edema; a similar process may occur at mucosal surfaces of the upper respiratory or gastrointestinal tract. Urticaria involves only the superficial portion of the dermis, presenting as well-circumscribed wheals with erythematous raised serpiginous borders and blanched centers that may coalesce to become giant wheals. Angioedema is a well-demarcated localized edema involving the deeper layers of the skin, including the subcutaneous tissue, and can also involve the bowel wall. Recurrent episodes of urticaria and/or angioedema of less than 6 weeks’ duration are considered acute, whereas attacks persisting beyond this period are designated chronic.
PREDISPOSING FACTORS AND ETIOLOGY
Urticaria and angioedema probably occur more frequently than reported because of the evanescent, self-limited nature of such eruptions, which seldom require medical attention when limited to the skin. Although persons in any age group may experience acute or chronic urticaria and/or angioedema, these lesions increase in frequency after adolescence, with the highest incidence occurring in persons in the third decade of life; indeed, one survey of college students indicated that 15–20% had experienced a pruritic wheal reaction.
The classification of urticaria-angioedema presented in Table 376-1 focuses on the different mechanisms for eliciting clinical disease and can be useful for differential diagnosis; nonetheless, most cases of chronic urticaria are idiopathic. Urticaria and/or angioedema occurring during the appropriate season in patients with seasonal respiratory allergy or as a result of exposure to animals or molds is attributed to inhalation or physical contact with pollens, animal dander, and mold spores, respectively. However, urticaria and angioedema secondary to inhalation are relatively uncommon compared to urticaria and angioedema elicited by ingestion of fresh fruits, shellfish, fish, milk products, chocolate, legumes including peanuts, and various drugs that may elicit not only the anaphylactic syndrome with prominent gastrointestinal complaints but also urticaria alone.
CLASSIFICATION OF URTICARIA AND/OR ANGIOEDEMA |
Additional etiologies include physical stimuli such as cold, heat, solar rays, exercise, and mechanical irritation. The physical urticarias can be distinguished by the precipitating event and other aspects of the clinical presentation. Dermographism, which occurs in 1–4% of the population, is defined by the appearance of a linear wheal at the site of a brisk stroke with a firm object or by any configuration appropriate to the eliciting event (Fig. 376-3). Dermographism has a prevalence that peaks in the second to third decades. It is not influenced by atopy and has a duration generally of <5 years. Pressure urticaria, which often accompanies chronic idiopathic urticaria, presents in response to a sustained stimulus such as a shoulder strap or belt, running (feet), or manual labor (hands). Cholinergic urticaria is distinctive in that the pruritic wheals are of small size (1–2 mm) and are surrounded by a large area of erythema; attacks are precipitated by fever, a hot bath or shower, or exercise and are presumptively attributed to a rise in core body temperature. Exercise-induced anaphylaxis can be precipitated by exertion alone or can be dependent on prior food ingestion. There is an association with the presence of IgE specific for α-5 gliadin, a component of wheat. The clinical presentation can be limited to flushing, erythema, and pruritic urticaria but may progress to angioedema of the face, oropharynx, larynx, or intestine or to vascular collapse; it is distinguished from cholinergic urticaria by presenting with wheals of conventional size and by not occurring with fever or a hot bath. Cold urticaria is local at body areas exposed to low ambient temperature or cold objects but can progress to vascular collapse with immersion in cold water (swimming). Solar urticaria is subdivided into six groups by the response to specific portions of the light spectrum. Vibratory angioedema may occur after years of occupational exposure or can be idiopathic; it may be accompanied by cholinergic urticaria. Other rare forms of physical allergy, always defined by stimulus-specific elicitation, include local heat urticaria, aquagenic urticaria from contact with water of any temperature (sometimes associated with polycythemia vera), and contact urticaria from direct interaction with some chemical substance.

FIGURE 376-3 Dermographic urticarial lesion induced by stroking the forearm lightly with the edge of a tongue blade. The photograph, taken after 2 minutes, demonstrates a prominent wheal-and-flare reaction in the shape of an X. (From LA Goldsmith et al [eds]: Fitzpatrick’s Dermatology in General Medicine, 8th ed. New York, McGraw-Hill, 2012. Photograph provided by Allen P. Kaplan, MD, Medical University of South Carolina.)
Angioedema without urticaria due to the generation of bradykinin occurs with C1 inhibitor (C1INH) deficiency that may be inborn as an autosomal dominant characteristic or may be acquired through the appearance of an autoantibody. The angiotensin-converting enzyme (ACE) inhibitors can provoke a similar clinical presentation in 0.1–0.5% of hypertensive patients due to attenuated degradation of bradykinin. The urticaria and angioedema associated with classic serum sickness or with hypocomplementemic cutaneous necrotizing angiitis are believed to be immune-complex diseases. The drug reactions to mast cell granule–releasing agents and to NSAIDs may be systemic, resembling anaphylaxis, or limited to cutaneous sites.
PATHOPHYSIOLOGY AND MANIFESTATIONS
Urticarial eruptions are distinctly pruritic, may involve any area of the body from the scalp to the soles of the feet, and appear in crops of 12- to 36-h duration, with old lesions fading as new ones appear. Most of the physical urticarias (cold, cholinergic, dermatographism) are an exception, with individual lesions lasting less than 2 h. The most common sites for urticaria are the extremities and face, with angioedema often being periorbital and in the lips. Although self-limited in duration, angioedema of the upper respiratory tract may be life-threatening due to laryngeal obstruction, whereas gastrointestinal involvement may present with abdominal colic, with or without nausea and vomiting, and may result in unnecessary surgical intervention. No residual discoloration occurs with either urticaria or angioedema unless there is an underlying vasculitic process leading to superimposed extravasation of erythrocytes.
The pathology is characterized by edema of the superficial dermis in urticaria and of the subcutaneous tissue and deep dermis in angioedema. Collagen bundles in affected areas are widely separated, and the venules are sometimes dilated. Any perivenular infiltrate consists of lymphocytes, monocytes, eosinophils, and neutrophils that are present in varying combination and numbers.
Perhaps the best-studied example of IgE- and mast cell–mediated urticaria and angioedema is cold urticaria. Cryoglobulins or cold agglutinins are present in up to 5% of these patients. Immersion of an extremity in an ice bath precipitates angioedema of the distal portion with urticaria at the air interface within minutes of the challenge. Histologic studies reveal marked mast cell degranulation with associated edema of the dermis and subcutaneous tissues. The histamine level in the plasma of venous effluent of the cold-challenged and angioedematous extremity is markedly increased, but no such increase appears in the plasma of effluent of the contralateral normal extremity. Elevated levels of histamine have been found in the plasma of venous effluent and in the fluid of suction blisters at experimentally induced lesional sites in patients with dermographism, pressure urticaria, vibratory angioedema, light urticaria, and heat urticaria. By ultrastructural analysis, the pattern of mast cell degranulation in cold urticaria resembles an IgE-mediated response with solubilization of granule contents, fusion of the perigranular and cell membranes, and discharge of granule contents, whereas in a dermographic lesion, there is additional superimposed zonal (piecemeal) degranulation. There are several reports of resolution of cold urticaria by treatment with monoclonal anti-human IgE (omalizumab). Elevations of plasma histamine levels with biopsyproven mast cell degranulation have also been demonstrated with generalized attacks of cholinergic urticaria and exercise-related anaphylaxis precipitated experimentally in subjects exercising on a treadmill while wearing a wet suit; however, only subjects with cholinergic urticaria have a concomitant decrease in pulmonary function.
Up to 40% of patients with chronic urticaria have an autoimmune cause for their disease including autoantibodies to IgE (5–10%) or, more commonly, to the α chain of FcεRI (35–45%). In these patients, autologous serum injected into their own skin can induce a wheal-and-flare reaction involving mast cell activation. The presence of these antibodies can also be recognized by their capacity to release histamine or induce activation markers such as CD63 or CD203 on basophils. An association with antibodies to microsomal peroxidase and/or thyroglobulin has been observed often with clinically significant Hashimoto’s thyroiditis. In vitro studies reveal that these autoantibodies can mediate basophil degranulation with enhancement by serum as a source of the anaphylatoxic fragment, C5a.
Hereditary angioedema is an autosomal dominant disease due to a deficiency of C1INH (type 1) in about 85% of patients and to a dysfunctional protein (type 2) in the remainder. A third type of hereditary angioedema has been described in which C1INH function is normal, and the causal lesion is a mutant form of factor XII, which leads to generation of excessive bradykinin. In the acquired form of C1INH deficiency, there is excessive consumption due either to immune complexes formed between anti-idiotypic antibody and monoclonal IgG presented by B cell lymphomas or to an autoantibody directed to C1INH. C1INH blocks the catalytic function of activated factor XII (Hageman factor) and of kallikrein, as well as the C1r/C1s components of C1. During clinical attacks of angioedema, C1INH-deficient patients have elevated plasma levels of bradykinin, particularly in the venous effluent of an involved extremity, and reduced levels of prekallikrein and high-molecular-weight kininogen, from which bradykinin is cleaved. The parallel decline in the complement substrates C4 and C2 reflects the action of activated C1 during such attacks. Mice with targeted disruption of the gene for C1INH exhibit a chronic increase in vascular permeability. The pathobiology is aggravated by administration of an ACE inhibitor (captopril) and is attenuated by breeding the C1INH null strain to a bradykinin 2 receptor (Bk2R) null strain. As ACE is also described as kininase II, the use of blockers results in impaired bradykinin degradation and explains the angioedema that occurs idiosyncratically in hypertensive patients with a normal C1INH. Bradykinin-mediated angioedema, whether caused by ACE inhibitors or by C1INH deficiency, is noteworthy for the conspicuous absence of concomitant urticaria.
DIAGNOSIS
The rapid onset and self-limited nature of urticarial and angioedematous eruptions are distinguishing features. Additional characteristics are the occurrence of the urticarial crops in various stages of evolution and the asymmetric distribution of the angioedema. Urticaria and/or angioedema involving IgE-dependent mechanisms are often appreciated by historic considerations implicating specific allergens or physical stimuli, by seasonal incidence, and by exposure to certain environments. Direct reproduction of the lesion with physical stimuli is particularly valuable because it so often establishes the cause of the lesion. The diagnosis of an environmental allergen based on the clinical history can be confirmed by skin testing or assay for allergen-specific IgE in serum. IgE-mediated urticaria and/or angioedema may or may not be associated with an elevation of total IgE or with peripheral eosinophilia. Fever, leukocytosis, and an elevated sedimentation rate are absent.
The classification of urticarial and angioedematous states presented in Table 376-1 in terms of possible mechanisms necessarily includes some differential diagnostic points. Hypocomplementemia is not observed in IgE-mediated mast cell disease and may reflect either an acquired abnormality generally attributed to the formation of immune complexes or a genetic or acquired deficiency of C1INH. Chronic recurrent urticaria, generally in females, associated with arthralgias, an elevated sedimentation rate, and normo- or hypocomplementemia suggests an underlying cutaneous necrotizing angiitis. Vasculitic urticaria typically persists longer than 72 h, whereas conventional urticaria often has a duration of 12–36 h. Confirmation depends on a biopsy that reveals cellular infiltration, nuclear debris, and fibrinoid necrosis of the venules. The same pathobiologic process accounts for the urticaria in association with such diseases as systemic lupus erythematosus or viral hepatitis with or without associated arteritis. Serum sickness per se or a similar clinical entity due to drugs includes not only urticaria but also pyrexia, lymphadenopathy, myalgia, and arthralgia or arthritis. Urticarial reactions to blood products or intravenous administration of immunoglobulin are defined by the event and generally are not progressive unless the recipient is IgA-deficient in the former case or the reagent is aggregated in the latter.
The diagnosis of hereditary angioedema is suggested not only by family history but also by the lack of pruritus and of urticarial lesions, the prominence of recurrent gastrointestinal attacks of colic, and episodes of laryngeal edema. Laboratory diagnosis depends on demonstrating a deficiency of C1INH antigen (type 1) or a nonfunctional protein (type 2) by a catalytic inhibition assay. While levels of C1 are normal, its substrates, C4 and C2, are chronically depleted and fall further during attacks due to the activation of additional C1. Patients with the acquired forms of C1INH deficiency have the same clinical manifestations but differ in the lack of a familial element. Furthermore, their sera exhibit a reduction of C1 function and C1q protein as well as C1INH, C4, and C2. Inborn C1INH deficiency and ACE inhibitor–elicited angioedema are associated with elevated levels of bradykinin. Lastly, type 3 hereditary angioedema is associated with normal levels of complement proteins.
Urticaria and angioedema are distinct from contact sensitivity, a vesicular eruption that progresses to chronic thickening of the skin with continued allergenic exposure. They also differ from atopic dermatitis, a condition that may present as erythema, edema, papules, vesiculation, and oozing proceeding to a subacute and chronic stage in which vesiculation is less marked or absent and scaling, fissuring, and lichenification predominate in a distribution that characteristically involves the flexor surfaces. In cutaneous mastocytosis, the reddish brown macules and papules, characteristic of urticaria pigmentosa, urticate with pruritus upon trauma; and in systemic mastocytosis, without or with urticaria pigmentosa, there is episodic systemic flushing with or without urtication but no angioedema.
SYSTEMIC MASTOCYTOSIS
DEFINITION
Systemic mastocytosis is defined by a clonal expansion of mast cells that in most instances is indolent and nonmalignant. The mast cell expansion is generally recognized only in bone marrow and in the normal peripheral distribution sites of the cells, such as skin, gastrointestinal mucosa, liver, and spleen. Mastocytosis occurs at any age and has a slight preponderance in males. The prevalence of systemic mastocytosis is not known, a familial occurrence is rare, and atopy is not increased.
CLASSIFICATION AND PATHOPHYSIOLOGY
A consensus classification for mastocytosis recognizes cutaneous mastocytosis with variants and four systemic forms (Table 376-2). Cutaneous mastocytosis is the most common diagnosis in children, whereas the form designated as indolent systemic mastocytosis (ISM) accounts for the majority of adult patients; it implies that there is no evidence of an associated hematologic disorder, liver disease, or lymphadenopathy and is not known to alter life expectancy. In systemic mastocytosis associated with clonal hematologic non–mast cell lineage disease (SM-AHNMD), the prognosis is determined by the nature of the associated disorder, which can range from dysmyelopoiesis to leukemia. In aggressive systemic mastocytosis (ASM), mast cell infiltration/proliferation in multiple organs such as liver, spleen, gut, and/or bone results in a poor prognosis; a subset of patients with this form has prominent eosinophilia with hepatosplenomegaly and lymphadenopathy. Mast cell leukemia (MCL) is the rarest form of the disease and is invariably fatal at present; the peripheral blood contains circulating, metachromatically staining, atypical mast cells. An aleukemic form of MCL is recognized without circulating mast cells when the percentage of high-grade immature mast cells in bone marrow smears exceeds 20% in a nonspicular area. Mast cell sarcoma and extracutaneous mastocytomas are rare solid mast cell tumors with malignant and benign features, respectively.
CLASSIFICATION OF MASTOCYTOSIS |
Cutaneous mastocytosis (CM)
Urticaria pigmentosa (UP)/maculopapular cutaneous mastocytosis (MPCM)
Variants: plaque form, nodular form; telangiectasia macularis eruptiva perstans (TMEP)
Solitary mastocytoma of skin
Diffuse cutaneous mastocytosis
Indolent systemic mastocytosis (ISM)
Systemic mastocytosis with an associated clonal hematologic non–mast cell lineage disease (SM-AHNMD)
Aggressive systemic mastocytosis (ASM)
Mast cell leukemia (MCL)
Mast cell sarcoma (MCS)
Extracutaneous mastocytoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


