Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections
SULFONAMIDES
The sulfonamide drugs were the first effective chemotherapeutic agents to be employed systemically for the prevention and cure of bacterial infections in humans. The advent of penicillin and other antibiotics diminished the usefulness of the sulfonamides, but the introduction of the combination of trimethoprim and sulfamethoxazole has increased the use of sulfonamides for the prophylaxis and treatment of specific microbial infections. Sulfonamides are derivatives of para– aminobenzenesulfonamide (sulfanilamide; Figure 52–1) and are congeners of para-aminobenzoic acid. Most of them are relatively insoluble in water, but their sodium salts are readily soluble.
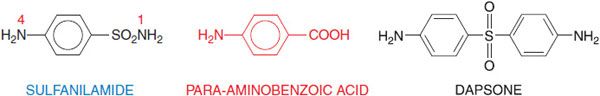
Figure 52–1 Sulfanilamide and para-aminobenzoic acid. Sulfonamides are derivatives of sulfanilamide and act by virtue of being congeners of para-aminobenzoate (PABA). The antimicrobial and dermatological anti-inflammatory agent dapsone (4,4’-diaminodiphenyl sulfone; see Figure 56–5 and Chapter 65) also bears a resemblance to PABA and sulfanilamide.
The minimal structural prerequisites for antibacterial action are all embodied in sulfanilamide itself. The sulfur must be linked directly to the benzene ring. The para-NH2 group (the N of which has been designated as N4) is essential and can be replaced only by moieties that can be converted in vivo to a free amino group. Substitutions made in the amide NH2 group (position N1) have variable effects on antibacterial activity of the molecule; substitution of heterocyclic aromatic nuclei at N1 yields highly potent compounds.
MECHANISM OF ACTION. Sulfonamides are competitive inhibitors of dihydropteroate synthase, the bacterial enzyme responsible for the incorporation of para-aminobenzoic acid (PABA) into dihydropteroic acid, the immediate precursor of folic acid (Figure 52–2). Sensitive microorganisms are those that must synthesize their own folic acid; bacteria that can use preformed folate are not affected. Toxicity is selective for bacteria because mammalian cells require preformed folic acid, cannot synthesize it, and are thus insensitive to drugs acting by this mechanism.
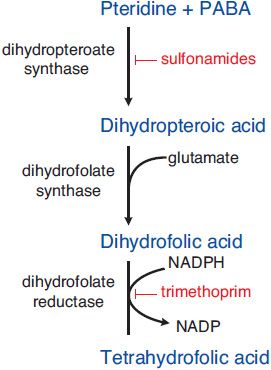
Figure 52–2 Steps in folate metabolism blocked by sulfonamides and trimethoprim. Coadministration of a sulfonamide and trimethoprim introduces sequential blocks in the biosynthetic pathway for tetrahydrofolate; the combination is much more effective than either agent alone.
SYNERGISTS OF SULFONAMIDES. Trimethoprim exerts a synergistic effect with sulfonamides. It is a potent and selective competitive inhibitor of microbial dihydrofolate reductase, the enzyme that reduces dihydrofolate to tetrahydrofolate, which is required for 1-carbon transfer reactions. Coadministration of a sulfonamide and trimethoprim introduces sequential blocks in the biosynthetic pathway for tetrahydrofolate (see Figure 52–2); the combination is much more effective than either agent alone.
EFFECTS ON MICROBES
Sulfonamides have a wide range of antimicrobial activity against both gram-positive and gram-negative bacteria. Resistant strains have become common and the usefulness of these agents has diminished correspondingly. Sulfonamides are bacteriostatic; cellular and humoral defense mechanisms of the host are essential for final eradication of the infection.
ANTIBACTERIAL SPECTRUM. Resistance to sulfonamides is increasingly a problem. Microorganisms that may be susceptible in vitro to sulfonamides include Streptococcus pyogenes, Streptococcus pneumoniae, Haemophilus influenzae, Haemophilus ducreyi, Nocardia, Actinomyces, Calymmatobacterium granulomatis, and Chlamydia trachomatis. Minimal inhibitory concentrations (MICs) range from 0.1 µg/mL for C. trachomatis to 4-64 µg/mL for Escherichia coli. Peak plasma drug concentrations achievable in vivo are ~100-200 µg/mL. Isolates of Neisseria meningitidis and Shigella are generally resistant, as are many strains of E. coli isolated from patients with urinary tract infections (community acquired).
ACQUIRED BACTERIAL RESISTANCE TO SULFONAMIDES. Bacterial resistance to sulfonamides can originate by random mutation and selection or by transfer of resistance by plasmids (see Chapter 48); it usually does not involve cross-resistance to other classes of antibiotics. Resistance to sulfonamide can result from: (1) a lower affinity of dihydropteroate synthase for sulfonamides, (2) decreased bacterial permeability or active efflux of the drug, (3) an alternative metabolic pathway for synthesis of an essential metabolite, or (4) an increased production of an essential metabolite or drug antagonist (e.g., PABA). Plasmid-mediated resistance is due to plasmid-encoded drug-resistant dihydropteroate synthetase.
ABSORPTION, DISTRIBUTION, METABOLISM, AND EXCRETION
Except for sulfonamides especially designed for their local effects in the bowel (see Chapter 47), this class of drugs is absorbed rapidly from the GI tract. Approximately 70-100% of an oral dose is absorbed, and sulfonamide can be found in the urine within 30 min of ingestion. Peak plasma levels are achieved in 2-6 h, depending on the drug. The small intestine is the major site of absorption, but some of the drug is absorbed from the stomach. Absorption from other sites, such as the vagina, respiratory tract, or abraded skin, is variable and unreliable, but a sufficient amount may enter the body to cause toxic reactions in susceptible persons or to produce sensitization.
All sulfonamides are bound in varying degree to plasma proteins, particularly to albumin. This is determined by the hydrophobicity of a particular drug and its pKa; at physiological pH, drugs with a high pKa exhibit a low degree of protein binding, and vice versa. Sulfonamides are distributed throughout all tissues of the body. The diffusible fraction of sulfadiazine is distributed uniformly throughout the total-body water, whereas sulfisoxazole is confined largely to the extracellular space. Because the protein content of such body fluids usually is low, the drug is present in the unbound active form. After systemic administration of adequate doses, sulfadiazine and sulfisoxazole attain concentrations in CSF that may be effective in meningitis. However, because of the emergence of sulfonamide-resistant microorganisms, these drugs are used rarely for the treatment of meningitis. Sulfonamides pass readily through the placenta and reach the fetal circulation. The concentrations attained in the fetal tissues may cause both antibacterial and toxic effects.
Sulfonamides are metabolized in the liver. The major metabolite is the N4-acetylated sulfonamide. Acetylation results in products that have no antibacterial activity but retain the toxic potential of the parent substance. Sulfonamides are eliminated from the body partly as the unchanged drug and partly as metabolic products. The largest fraction is excreted in the urine, and the t1/2 depends on renal function. In acid urine, the older sulfonamides are insoluble and crystalline deposits may form. Small amounts are eliminated in the feces, bile, milk, and other secretions.
PHARMACOLOGICAL PROPERTIES OF INDIVIDUAL SULFONAMIDES
The sulfonamides may be classified on the basis of the rapidity with which they are absorbed and excreted (Table 52–1).
Table 52–1
Classes of Sulfonamides
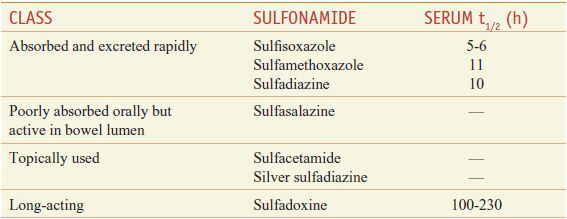
RAPIDLY ABSORBED AND ELIMINATED SULFONAMIDES
Sulfisoxazole. Sulfisoxazole is a rapidly absorbed and excreted sulfonamide with excellent antibacterial activity. Its high solubility eliminates much of the renal toxicity inherent in the use of older sulfonamides. Sulfisoxazole is bound extensively to plasma proteins. Following an oral dose of 2-4 g, peak concentrations in plasma of 110-250 µg/mL are found in 2-4 h. Approximately 30% of sulfisoxazole in the blood and ~30% in the urine is in the acetylated form. The kidney excretes ~95% of a single dose in 24 h. Concentrations of the drug in urine thus greatly exceed those in blood and may be bactericidal. The concentration in CSF is about a third of that in the blood. Sulfisoxazole acetyl is marketed in combination with erythromycin ethylsuccinate for use in children with otitis media.
The untoward effects of sulfisoxazole are similar to other sulfonamides. Because of its relatively high solubility in the urine as compared with sulfadiazine, sulfisoxazole rarely produces hematuria or crystalluria (0.2-0.3%). Nonetheless, patients should still ingest an adequate quantity of water. Sulfisoxazole must be used with caution in patients with impaired renal function. Like all sulfonamides, sulfisoxazole may produce hypersensitivity reactions, some of which are potentially lethal.
Sulfamethoxazole. Sulfamethoxazole is a close congener of sulfisoxazole, but its rates of enteric absorption and urinary excretion are slower. It is administered orally and employed for both systemic and urinary tract infections. Precautions must be observed to avoid sulfamethoxazole crystalluria because of the high percentage of the acetylated, relatively insoluble form of the drug in the urine. The clinical uses of sulfamethoxazole are the same as those for sulfisoxazole. In the U.S., it is marketed only in fixed-dose combinations with trimethoprim.
Sulfadiazine. Sulfadiazine given orally is absorbed rapidly from the GI tract. Peak blood concentrations are reached within 3-6 h. About 55% of the drug is bound to plasma protein. Therapeutic concentrations are attained in CSF within 4 h of a single oral dose of 60 mg/kg. Both free and acetylated forms of sulfadiazine are readily excreted by the kidney; 15-40% of the excreted drug is in acetylated form. Alkalinization of the urine accelerates the renal clearance of both forms by diminishing their tubular reabsorption. Every precaution must be taken to ensure fluid intake adequate to produce a daily urine output of at least 1200 mL in adults and a corresponding quantity in children. If this cannot be accomplished, sodium bicarbonate may be given to reduce the risk of crystalluria.
POORLY ABSORBED SULFONAMIDES
SULFASALAZINE. Sulfasalazine (AZULFIDINE, others) is very poorly absorbed from the GI tract. It is used in the therapy of ulcerative colitis and regional enteritis. Intestinal bacteria break sulfasalazine down to 5-aminosalicylate (5-ASA, mesalamine; see Figures 47–2 through 47–4), the active agent in inflammatory bowel disease, and sulfapyridine, a sulfonamide that is absorbed and eventually excreted in the urine.
SULFONAMIDES FOR TOPICAL USE
Sulfacetamide. Sulfacetamide is the N1-acetyl-substituted derivative of sulfanilamide. Its aqueous solubility is ~90 times that of sulfadiazine. Solutions of the sodium salt of the drug are employed extensively in the management of ophthalmic infections. Very high aqueous concentrations are not irritating to the eye and are effective against susceptible microorganisms. The drug penetrates into ocular fluids and tissues in high concentration. Sensitivity reactions to sulfacetamide are rare, but the drug should not be used in patients with known hypersensitivity to sulfonamides. A 30% solution of the sodium salt has a pH of 7.4, whereas the solutions of sodium salts of other sulfonamides are highly alkaline. See Chapters 64 and 65 for ocular and dermatological uses.
Silver Sulfadiazine. Silver sulfadiazine (SILVADENE, others) is used topically to reduce microbial colonization and the incidence of infections from burns. Silver sulfadiazine should not be used to treat an established deep infection. Silver is released slowly from the preparation in concentrations that are selectively toxic to the microorganisms. However, bacteria may develop resistance to silver sulfadiazine. Although little silver is absorbed, the plasma concentration of sulfadiazine may approach therapeutic levels if a large surface area is involved. Adverse reactions—burning, rash, and itching—are infrequent. Silver sulfadiazine is considered an agent of choice for the prevention of burn infections.
Mafenide. This sulfonamide (α-amino-p-toluene-sulfonamide; SULFAMYLON) is applied topically to prevent colonization of burns by a large variety of gram-negative and gram-positive bacteria. It should not be used in treatment of an established deep infection. Adverse effects include intense pain at sites of application, allergic reactions, and loss of fluid by evaporation from the burn surface because occlusive dressings are not used. The drug and its primary metabolite inhibit carbonic anhydrase, and the urine becomes alkaline. Metabolic acidosis with compensatory tachypnea and hyperventilation may ensue; these effects limit the usefulness of mafenide. Mafenide is absorbed from the burn surface, reaching peak plasma concentrations in 2-4 h; the absorbed drug is converted to para-carboxybenzenesulfonamide.
LONG-ACTING SULFONAMIDES
Sulfadoxine. This agent has a particularly long plasma t1/2 (7-9 days). It is used in combination with pyrimethamine (500 mg sulfadoxine plus 25 mg pyrimethamine as FANSIDAR) for the prophylaxis and treatment of malaria caused by mefloquine-resistant strains of Plasmodium falciparum (see Chapter 49). However, because of severe and sometimes fatal reactions, including the Stevens-Johnson syndrome, and the emergence of resistant strains, the drug has limited usefulness for the treatment of malaria.
SULFONAMIDE THERAPY
URINARY TRACT INFECTIONS (UTIs). Because a significant percentage of UTIs are caused by sulfonamide-resistant microorganisms, sulfonamides are no longer a therapy of first choice. Trimethoprim-sulfamethoxazole, a quinolone, trimethoprim, fosfomycin, or ampicillin are the preferred agents. Sulfisoxazole may be used effectively in areas where the prevalence of resistance is not high. The usual dosage is 2-4 g initially followed by 1-2 g, orally 4 times a day for 5-10 days. Patients with acute pyelonephritis with high fever are at risk of bacteremia and shock and should not be treated with a sulfonamide.
NOCARDIOSIS. Sulfonamides are of value in the treatment of infections due to Nocardia spp. Sulfisoxazole or sulfadiazine may be given in dosages of 6-8 g daily and this schedule should be continued for several months after all manifestations have been controlled. Combination of sulfonamide with a second antibiotic has been recommended, especially for advanced cases; ampicillin, erythromycin, and streptomycin have been suggested but there are no clinical data to show that combination therapy is better than therapy with a sulfonamide alone. Trimethoprim-sulfamethoxazole also is effective; some authorities consider this combination to be the treatment of choice.
TOXOPLASMOSIS. The combination of pyrimethamine and sulfadiazine is the treatment of choice for toxoplasmosis (see Chapter 50). Pyrimethamine is given as a loading dose of 75 mg followed by 25 mg orally per day, with sulfadiazine 1 g orally every 6 h, plus folinic acid (leucovorin) 10 mg orally each day for at least 3-6 weeks. Patients should receive at least 2 L of fluid intake daily to prevent crystalluria.
USE OF SULFONAMIDES FOR PROPHYLAXIS. The sulfonamides are as efficacious as oral penicillin in preventing streptococcal infections and recurrences of rheumatic fever among susceptible subjects. Their toxicity and the possibility of infection by drug-resistant streptococci make sulfonamides less desirable than penicillin for this purpose: however, sulfonamides should be used in patients who are hypersensitive to penicillin. Untoward responses usually occur during the first 8 weeks of therapy. White blood cell counts should be carried out once weekly during the first 8 weeks.
UNTOWARD REACTIONS TO SULFONAMIDES
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


