Table 3.1.1 Therapeutic indications for the sulfonamide antibiotics.
| Sulfonamide antibiotic | Indications |
| Co-trimoxazole (sulfamethoxazole and trimethoprim) | Treatment prevention of Pneumocystis jirovecii pneumonia |
| Treatment of toxoplasmosis | |
| Treatment of nocardiosis | |
| Sulfadiazine | Prevention of rheumatic fever |
| Treatment of toxoplasmosis | |
| Prevention and treatment of infection in burn wounds (used topically as the silver sulfadiazine salt) |
3.1.1 Discovery
Although there are only two sulfonamides currently in clinical use, and their indications are very specific, this class of drugs deserves detailed study as the sulfonamides were the first specific and synthetic antibacterials and, as we shall see, they have also helped shape the course of history. The sulfonamides were discovered in 1932 when a red azo dye (Prontosil), synthesised by the chemist Josef Klarer at the Bayer Corporation (IG Farbenindustrie), was found to prevent the multiplication of bacteria in animal experiments (but not in in vitro tests). Heinrich Hörlein had initiated a pharmaceutical research programme in the 1920s which was aimed at the treatment of bacterial and tropical diseases with synthetic chemicals, and by the end of the decade, more than 30 new synthetic chemicals were being tested per week against a streptococcal strain (Mietzsch and Klarer, 1935; Bentley, 2009).
Gerhard Domagk, who was responsible for the antibacterial testing and discovered the antibacterial effect of the sulfonamides (Domagk, 1935), was awarded the Nobel Prize in Medicine in 1939, but was forbidden from accepting the prize by Adolf Hitler (http://nobelprize.org/nobel_prizes/medicine/laureates/1939/domagk-lecture.pdf, last accessed 29 March 2012). After the war, Domagk was able to accept the prize, but was not given the monetary sum which went with it, as this was said to have expired (this is the kind of bad luck we can sympathise with) (Sköld, 2000).
As the authors can confirm, having taught on a medicinal chemistry module which involved their synthesis and analysis, the sulfonamides are not the most soluble of drugs, and an early attempt (1937) by Harold Cole Watkins to prepare a soluble form resulted in the tragic deaths of more than 100 patients who had taken Elixir Sulfanilamide1 (Figure 3.1.2), as diethylene glycol (HOCH2CH2OCH2CH2OH), a highly toxic chemical which is used as an antifreeze, had been used as the solvent. This tragedy ultimately led to the increased regulation of drugs by the US Food and Drug Administration (FDA) (Wax, 1995).
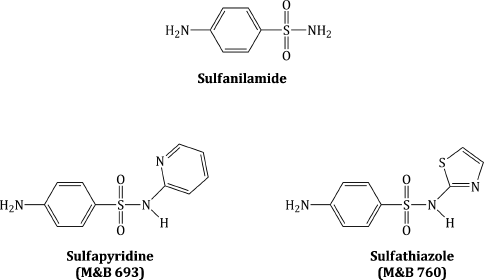
On a more positive historical note, a sulfonamide, thought to be either sulfapyridine (then known as M&B 693)2 or sulfathiazole (M&B 760) (Figure 3.1.2), was used to treat Winston Churchill, who had contracted pneumonia in 1943, en route to Tunisia after a tour of the Middle East to meet Allied leaders. Churchill’s personal physician, Lord Moran, feared for the life of the Prime Minister (and the effect that his death would have on the morale of the British nation) and called in Brigadier D. E. Bedford, a heart specialist, as Churchill was also having heart fibrillations, thus giving Churchill his opportunity to joke that ‘The M&B, which I may also call Moran and Bedford, did the job most effectively. There is no doubt that pneumonia is a very different illness from what it was before this marvellous drug was discovered’ (Lesch, 2007).
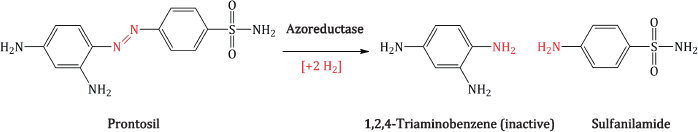
Anyway, that’s enough history for now; let’s get back to the discovery of the sulfonamides. Prontosil was soon found to be inactive in vitro, as it is a prodrug which requires activation, via an azoreductase enzyme in vivo (Scheme 3.1.1). Using prontosil, Domagk was able to control streptococcal infections in mice and staphylococcal infections in rabbits. In 1935, Ernest Fourneau and his colleagues at the Institute Pasteur in Paris discovered the in vivo metabolism of prontosil to sulfanilamide, which soon became the first synthetic antibacterial agent and was on the market in the UK, France, and USA by 1937 (Tréfouël et al., 1935).
3.1.2 Synthesis
Having discovered this exciting antibacterial activity, the research groups mentioned above were joined by pharmaceutical companies from all over the world, which began to synthesise sulfanilamide analogues, initially as probes of the structural requirements for activity, thereafter in attempts to prepare sulfonamides with more desirable properties: increased antibacterial activity, a broader spectrum of activity, reduced toxicity, longer half-life, and so on. Many thousands of analogues were tested and the sulfonamide group (-SO2NR2) and a free amino group at the para-position of an otherwise unsubstituted benzene ring were found to be essential (Figure 3.1.3) (Domagk, 1957; Miller et al., 1972).
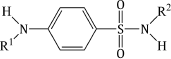
In summary, only variations in R1 (on N1) and R4 (on N4) lead to active sulfonamides; R4 must be a group that can be hydrolysed in vivo to give the active free amino group, while variations in R1 (especially the introduction of heterocyclic substituents) are responsible for the many sulfonamides which have been used clinically. Thankfully, there are a number of possible synthetic routes to suitably substituted sulfonamides, and we will mention two here.
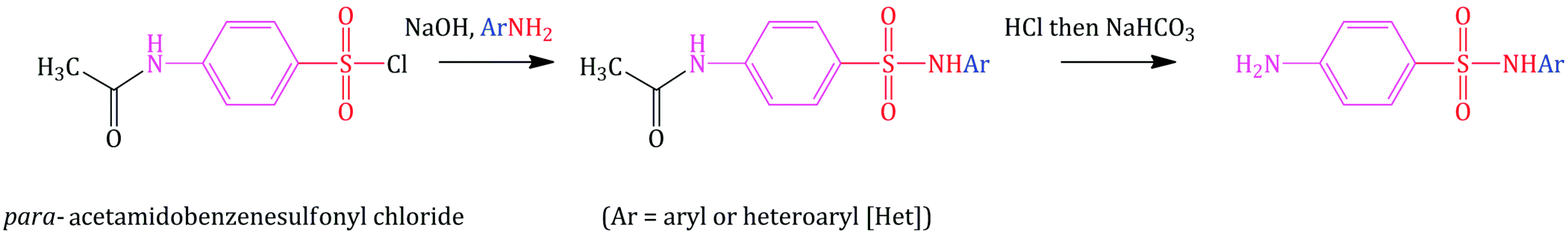
A wonderfully simple looking route involves the nucleophilic substitution of commercially available p-acetamidobenzenesulfonyl chloride by an aromatic amine, followed by hydrolysis of the intermediate (Scheme 3.1.2) (Winnek and Roblin, 1947). The acetyl group in the starting material is being used as a protecting group, as it prevents any unwanted side reactions of the amino group and is hydrolysed in the second step to release the biologically active free amine. In theory, this route could be used to prepare any heteroaryl-substituted sulfonamide, as there are any number of aromatic amines which could be used in the nucleophilic substitution step. In practice, however, this route is limited by the solubilities of the intermediates and by the final products in the aqueous acidic, then basic, conditions employed in the hydrolysis step.
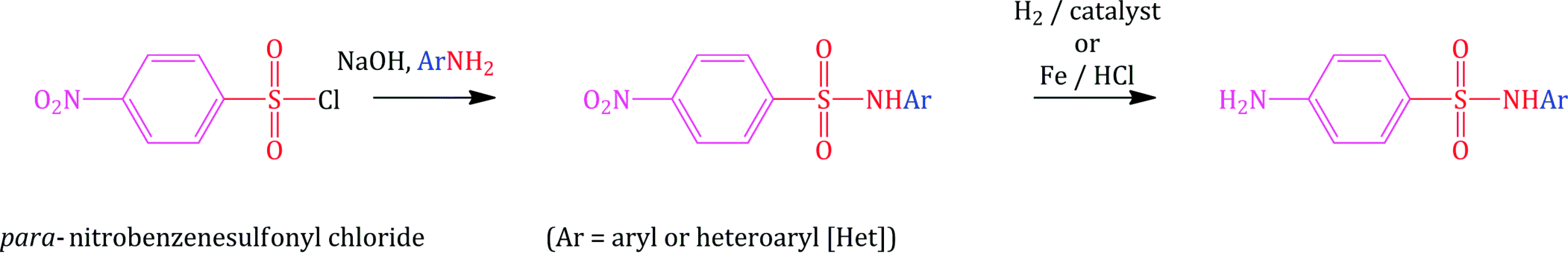
An equally attractive method involves the initial nucleophilic substitution of p-nitrobenzenesulfonyl chloride (Scheme 3.1.3), in which the nitro group cannot undergo any unwanted side reactions and will also facilitate the initial substitution through its strongly electron-withdrawing effect. In this case, the free amino group can be generated in the second step by reduction of the aromatic nitro group in a suitable solvent, a well-studied synthetic process (Senear et al., 1946; Luk’yanov et al., 1980).
Scheme 3.1.3 Synthesis of sulfonamides via nucleophilic substitution followed by nitro-group reduction
3.1.3 Bioavailability
The oral bioavailability of the individual sulfonamides is not affected by co-administration with food, but, although generally good, it is dependent upon the acidity of the sulfonamide (pKa),3 as this determines the degree of ionisation at the various pHs of the GI tract and, consequently, the lipophilicity and absorption across the intestinal wall.
The general structure of a sulfonamide antibacterial agent is shown in Scheme 3.1.4. If you consider the structure, you can identify two sites that can take part in acid–base equilibria: the aromatic amine (pKa1), which has low basicity due to resonance of the electron pair with the aromatic system and through to the sulfonyl group, and the sulfonamide NH (pKa2), which is acidic, due to stabilisation of the conjugate anion by delocalisation with both the sulfonyl group and the heterocyclic substituent. The basicity of the aromatic amine is sufficiently low that it is poorly protonated, even in the stomach, and it varies little throughout the sulfonamide series, since, as we discovered in the previous subsection, the introduction of substituents on to the aromatic ring (which would affect the basicity) results in a loss of antibacterial activity. The acidity of the sulfonamide NH is more variable as it is dependent upon the nature of the heterocyclic substituent, as indicated in Table 3.1.2 (Hekster and Vree, 1982). It is sufficiently acidic in the majority of sulfonamides that many exist predominantly as the anion in most physiological compartments, which aids with their solubility and distribution.
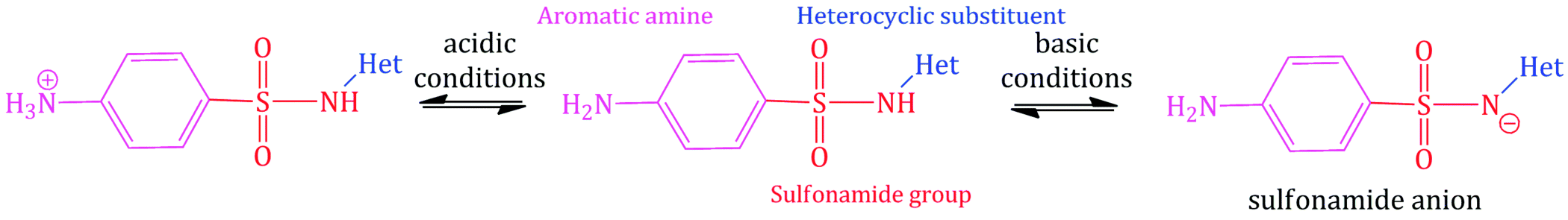
Table 3.1.2 pKa values for some common sulfonamides (HA in parentheses).
| Sulfonamide | pKavalues | Reference |
| Sulfamethoxazole | pKa1 (NH3+) 1.85, pKa2 (NH) 5.60 | Qiang and Adams (2004) |
| Sulfadiazine | pKa1 (NH3+) 2.10, pKa2 (NH) 6.28 | Lin et al. (1997) |
| Sulfathiazole | pKa1 (NH3+) 2.01, pKa2 (NH) 7.11 | Qiang and Adams (2004) |
| Sulfadoxine | pKa1 (NH3+) 1.59, pKa2 (NH) 5.82 | Ghafourian et al. (2006) |
The solubility of a molecule is affected by its inherent solubility, taking into account the number and nature of polar groups and the extent of ionisation at any given pH. The oral bioavailability is affected by the solubility, because some aqueous solubility is required in the GI tract in order to allow the sulfonamide to partition between the aqueous gastric contents and the lipophilic GI wall.
In the blood, those sulfonamides with a pKa2 value below the blood pH will be ionised and exist as the sulfonamide anion and as a result will have good solubility and distribution, being well distributed throughout the body, with an apparent volume of distribution (Vd)4 of approximately 0.1–0.25 L/kg in adults (Ghafourian et al., 2006), reaching most tissues and compartments and conferring wide therapeutic application.
The aqueous solubility also affects the renal clearance of the sulfonamides; greater aqueous solubility, in general, increases the renal excretion. However, systemic sulfonamides with low aqueous solubility, such as sulfadiazine (see Table 3.1.3), can crystallise in the blood and kidneys, which causes irritation to the renal tubules. The co-administration of sodium bicarbonate results in a slight increase in the blood pH and an accompanying increase in sulfonamide solubility, reducing the risk of crystallisation (Hekster and Vree, 1982).
Table 3.1.3 The solubility of some sulfonamides at different pH values and selected pharmacokinetic data.

As a result of the variations in solubility, absorption, and excretion of the sulfonamides, they can be classified into separate groups for systemic use: ultrashort acting, short–medium acting, and long acting; as well as those for GI and topical use.
The oral sulfonamides commonly used clinically, sulfamethoxazole (in co-trimoxazole) and sulfadiazine, fall into the category of short–medium acting agents that are well absorbed, with reported bioavailabilities of 100% after oral administration (Bertz and Granneman, 1997) and blood plasma half-life (t½) values of 4–16 hours (Hekster and Vree, 1982). Peak plasma concentrations are seen within 1–3 hours after oral administration (Hekster and Vree, 1982). Long-acting agents, such as sulfadimethoxine, have been largely replaced by less-toxic sulfonamides, due to reports of severe allergic reactions (e.g. Rallison et al., 1961; reviewed in Choquet-Kastylevsky et al., 2002; Brackett 2007), not only in humans, but also in animals (e.g. Campbell, 1999; Trepanier, 1999). Sulfadoxine is an example of a long-acting sulfonamide (t½ about 110 hours) that remains in clinical use – in combination with pyrimethamine, it is indicated for the treatment of malaria and toxoplasmosis. Its long half-life is believed to be appropriate for the slow life cycle of Plasmodium falciparum, the causative bacterium in malaria.
GI sulfonamides are those with low absorption from the GI tract; such agents have been used in sterilising the GI tract before surgery, but this is now usually achieved through the use of aminoglycosides. Sulfapyridine is the only sulfonamide from this group still used clinically (in the treatment of ulcerative colitis), as part of the prodrug sulfasalazine, which is reduced by bacterial azoreductase in the lower GI tract to release 5-aminosalicylic acid (which is considered to be the active agent) and sulfapyridine. Sulfonamides have been used for the topical treatment of skin and wound infections for many years, since the effectiveness of the combination of sulfamethoxazole/trimethoprim was observed by three colonels and a major in the US Army Medical Corps (Moncrieff et al., 1966).
The relationship between pKa2 and the solubility of the sulfonamide is also important for uptake into the bacterial target – we will look at this in the last part of this subsection. The data in Table 3.1.3 show that the sulfonamides listed have greater aqueous solubility at pH 7 (when they are all ionised to some extent) than at pH 5.
Much work has gone into the study of the pharmacokinetic parameters of sulfonamides, especially in comparison to those of trimethoprim, to identify the optimal sulfonamide for combination therapy with trimethoprim (reviewed in Watson et al., 1988 and in Hekster and Vree, 1982). In designing a combination therapy, it is essential that the two (or more) individual agents have compatible pharmacokinetics in order to ensure that therapeutic concentrations at the target site(s) are achieved simultaneously. If the combination agents do not act together at the site of infection, the synergistic action is lost and the development of resistance becomes a significant risk. Even though the combination of sulfamethoxazole and trimethoprim has been used for many years, and continues to be a key treatment for certain pneumonial infections, the differences in their respective bioavailability and distribution have caused some discussion and even criticism of their combined use (O’Grady, 1975; Reeves and Wilkinson, 1979; Watson et al., 1981; Männistö et al., 1982; Burman, 1986; Królicki et al., 2004).
All of the current sulfonamide antibacterial agents are significantly metabolised to their corresponding N4-acetyl derivatives, through acetylation by N-acetyl transferase (NAT) activity, predominantly in the liver. The aromatic amine (N4) must be unsubstituted for antibacterial activity, so N4-acetylation leads to inactive metabolites, which are excreted in the urine, along with unchanged sulfonamide. Early pharmacogenetic analysis of NAT activity led to the classification of patients as ‘slow’ or ‘fast’ acetylators (Evans and White, 1964; Evans, 1969; Schroder and Evans, 1972); unfortunately, the rate of acetylation of each individual agent does not necessarily hold across the series of sulfonamides, preventing reliable clinical prediction (Hekster and Vree, 1982). For slow acetylators, there is the risk of toxicity due to increased levels of sulfonamide, while fast acetylators risk sub-therapeutic levels of the antibacterial agent and a less favourable response to treatment. Even in the early 1980s, the possibility of personalised medicine was raised in the context of sulfonamide treatment of bacterial infections (Vree et al., 1980).
Like their sulfonamide parent drugs, the N4-acetyl metabolites are more soluble at pH 7.0 than at pH 5.5, although they are usually less soluble than the sulfonamide parent. However, measurement and comparison of the urinary excretion of sulfonamides to that of their N4-acetyl metabolites has shown that the direct relationship between pH and sulfonamide excretion was not mirrored by the N4-acetyl compounds, which maintained a relatively constant urinary excretion that was almost independent of pH (this phenomenon is exemplified by the behaviour of sulfamethoxazole and N4-acetylsulfamethoxazole) (Vree et al., 1978). Urinary excretion accounts for about 90% or more of the route of elimination for sulfonamides and their N4-acetyl metabolites (Hekster and Vree, 1982).
Other minor metabolic pathways result in N1-glucuronidation and hydroxylation of the phenyl and/or heterocyclic rings. O– and N-dealkylation are also possible in appropriately substituted sulfonamides (Hekster and Vree, 1982).
3.1.3.1 Bacterial Uptake of Sulfonamides
The concentration of sulfonamide found inside a bacterial cell depends upon its ability to enter the cell (and be retained once inside), which depends upon the difference in pH between the extracellular medium and the cytoplasm of the bacterial cell, as this determines the extent of ionisation of the sulfonamide in each site (Tappe et al., 2008; Zarfl et al., 2008). In essence, sulfonamides with a pKa2 close to 5–7 are better accumulated in bacteria, since they predominantly exist in the unionised (neutral) form outside the cell, and can pass through the bacterial membrane by passive diffusion, then become partly ionised inside the bacterial cells, due to the slightly higher pH (usually around 7.6), leaving them trapped. Sulfonamides with a higher pKa2 value are ionised to a lesser extent in bacterial cells, as this would require more basic conditions than the intracellular pH provides; these sulfonamides can freely diffuse into and out of the cell, preventing their accumulation.
3.1.4 Mode of Action and Selectivity
During the early antibacterial studies, it was discovered that the sulfonamides (then referred to as sulphonamides, sulpha, or sulfa drugs) were bacteriostatic, as they prevented the growth of bacteria, but did not kill them, and that, when para-aminobenzoic acid (PABA) was added to bacterial culture at the same time as sulfonamide, the drug had little or no effect.
As we learned in Subsection 1.1.3.2, bacteria require PABA (an essential metabolite) for the synthesis of folic acid and lack the protein required for folate uptake from their environment. Folic acid is an essential metabolite for mammals as it cannot be synthesised by mammalian cells and must be obtained from the mammalian diet (you can check how much folic acid you require daily by reading the ingredients on the side of your breakfast cereal packet). The sulfonamides are thus highly selective as they interfere with a key process in bacterial (prokaryotic) cells which does not take place in eukaryotic (mammalian) cells.
We now know that this interference in folic acid synthesis is the basis of the selectivity of the sulfonamides, but why is folic acid so important? Folic acid is indirectly involved in DNA synthesis as the enzyme co-factors which are required for the synthesis of the purine and pyrimidine bases of DNA are derivatives of folic acid (see Subsection 1.1.3.2). If the synthesis of folic acid is inhibited, the cellular supply of these co-factors will be diminished, and DNA synthesis will be prevented.
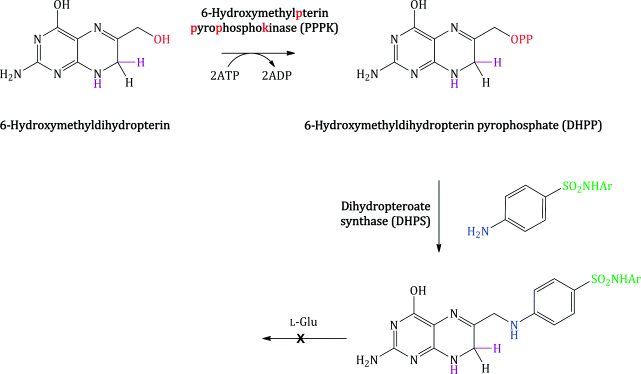
Bacterial synthesis of folic acid (actually dihydrofolic acid) involves a number of steps, with the key steps in terms of the mode of action of the sulfonamides shown in Scheme 3.1.5. Once again, a nucleophilic substitution is involved, in which the free amino group of PABA substitutes for the pyrophosphate group introduced on to 6-hydroxymethylpterin by the enzyme PPPK. In the final step, amide formation takes place between the free amino group of L-glutamic acid and the carboxylic acid group derived from PABA (Achari et al., 1997).
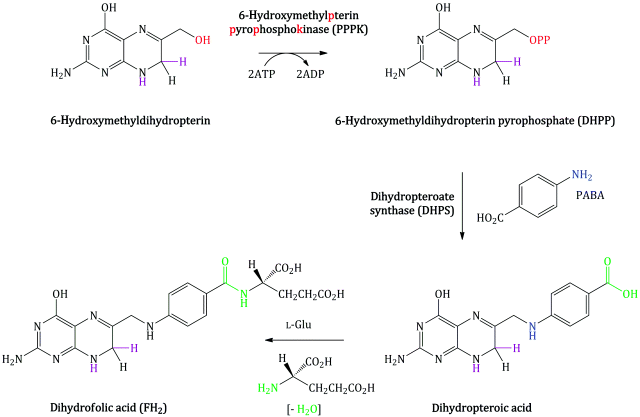
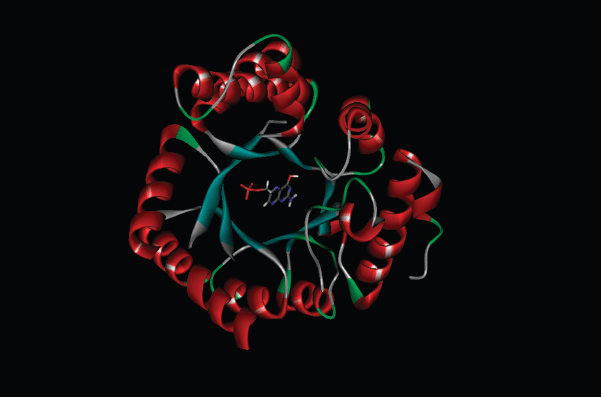
Sulfanilamide and suitably substituted derivatives have similar molecular dimensions and properties to PABA (Figure 3.1.4), so are mistaken for it by the enzyme dihydropteroate synthase. These sulfonamides bind competitively to the PABA binding domain of the active site of dihydropteroate synthase (which is highly conserved between bacteria) (Figure 3.1.5), and some are incorporated into the macromolecule (Roland et al., 1979). Once incorporated into the macromolecule, further reaction with L-glutamic acid will not produce folic acid (Scheme 3.1.6). The incorporation of the sulfonamide is, however, reversible, since the addition of excess PABA results in the formation of folic acid (Woods, 1940).
Figure 3.1.4 (a) Comparison of molecular properties of PABA and sulfonamides. (b) The electrostatic similarities between PABA (top) and sulfamethoxazole (bottom)
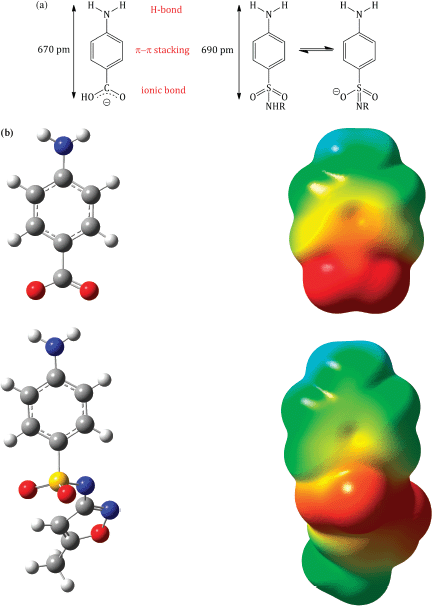
Figure 3.1.5 Dihydropteroate synthase (DHPS) from S. pneumoniae with bound DHPP; PDB code 2VEG (Levy et al., 2008)
You will already have noticed why the free amino group is required in the sulfonamides: the introduction of any substituent other than hydrogen may well interfere (electronically or sterically) with the nucleophilic substitution which is key to this mode of action.
The sulfonamides are thus examples of antimetabolites (more properly, antifolates), which are defined as agents that are similar in structure to an essential metabolite but cannot take its place. The antimetabolite theory attributed to Woods (1940) and Fildes (1940) is most applicable to the sulfonamides, trimethoprim, and the anticancer agents 5-fluorouracil, 6-mercaptopurine, and methotrexate.
Look out for more on the mode of action of the sulfonamides in Section 3.2, where we will learn about the dihydrofolate reductase inhibitor trimethoprim and the combination of sulfamethoxazole and trimethoprim (co-trimoxazole).
3.1.5 Bacterial Resistance
As we have said a number of times so far, knowing the mode of action of an antibacterial agent gives us an idea of how bacteria will develop resistance, and it should, therefore, come as no surprise that altering the structure of dihydropteroate synthase, the enzyme that the sulfonamides bind to (in competition with PABA), resulting in some of them being incorporated into the folic acid precursor, is one of the ways in which bacterial cells overcome the effects of these agents.
Chromosomal resistance through mutations in the dihydropteroate synthase (folP) gene in Escherichia coli, Staphylococcus aureus, Pneumocystis jirovecii (fungal pneumonia infection), Campylobacter jejuni (gastroenteritis), and Neisseria meningitidis (bacterial meningitis) leads to alterations in the sulfonamide binding site of the DHPS. In one such E. coli mutant, the result of a single base-pair mutation leading to a Phe changing to Ile at position 28 of the amino acid sequence, led to a trade-off between resistance and enzyme efficiency, with the Ki for sulfathiazole being increased by 150 times and the KM for PABA by 10 times5 (Dallas et al., 1992).
Sulfonamide resistance in Gram negative bacteria is plasmid-borne, with the two plasmid-borne genes sul1 and sul2, found in roughly equal frequency among clinical isolates, again encoding drug-resistant DHPS variants (Akiba et al., 1960; Sköld, 1976). For more on the bacterial resistance of the sulfonamides, see Sköld (2000).
3.1.6 Clinical Applications
3.1.6.1 Spectrum of Activity
The sulfonamides are broad-spectrum antibiotics, with activity against both Gram negative and Gram positive bacteria, ranging from Streptococcus pneumonia, Nocardia asteroides, E. coli, Brucella species, and Corynebacterium diphtheria, to the protozoan Toxoplasma gondii (Allegra et al., 1990). When first discovered, the sulfonamides were used to treat a wide range of clinical conditions, but nowadays, due to toxicity problems (e.g. blood dyscrasias and Stevens–Johnson syndrome) and increased resistance, they are only used as first-line treatment for a few clinical conditions (discussed below). The sulfonamides should only be used to treat common infections (such as urinary tract infections [UTIs], otitis media, and bronchitis) when there is good bacteriological evidence suggesting sensitivity and there is a clinical reason to use a sulfonamide in preference to another antibiotic. The sulfonamides lack activity against mycobacteria, while strains of Pseudomonas aeruginosa are often resistant (Poole, 2001; Livermore, 2002), and this limits their usefulness against diseases caused by these pathogens.
3.1.6.2 Protozoan Infections
As infections due to protozoa and fungi are outwith the scope of this book, we will mention these here only in brief. Toxoplasmosis is a widespread parasitic disease caused by the protozoan parasite Toxoplasma gondii. A common source of T. gondii infection is from cats, as T. gondii replicates in the intestines of cats and is expelled in cats’ faeces. As a result, any food or water ingested that is contaminated by cat faeces may result in the development of T. gondii infection (a good reason to keep cats off your vegetable patch) (Dabritz and Conrad, 2010).
When the patient has a healthy immune system, the infection does generally not cause any symptoms, but in cases where the immune system is compromised (such as in HIV), the infection can affect the brain, causing toxoplasmic encephalitis (TE), which can present as a multitude of neurological symptoms, including seizures, ataxia (loss of muscle coordination resulting in lack of balance), headaches, fever, and confusion. TE is an opportunistic infection, and if left untreated can be fatal, and it is a major cause of mortality in patients with HIV/AIDS, particularly in developing countries. Another scenario where T. gondii infection can be serious is if the infection is contracted during pregnancy, as T. gondii can cross the placenta to the foetus leading to foetal mental retardation and blindness, a condition known as congenital toxoplasmosis.
In those cases where infection with T. gondii can be serious (and result in complications), treatment can be prescribed to eradicate the parasite. Generally, the co-administration of sulfadiazine and pyrimethamine (an antimalarial drug which, like trimethoprim, is a DHFR inhibitor) is used as first-line treatment. Co-trimoxazole is used as an alternate therapy in resource-poor areas where sulfadiazine and pyrimethamine are not available. Treatment usually lasts for 3–6 weeks, but if the infection is associated with HIV/AIDS, the patient may have to take the medication for longer (sometimes for life) in order to keep it under control.
A Cochrane review failed to recommend any one regimen for the treatment of TE due to the lack of good-quality clinical trials, but from the data available, no significant differences in clinical response were found between co-trimoxazole alone and sulfadiazine and pyrimethamine together (Dedicoat and Livesley, 2006).
Pneumocystis jirovecii (formally known as Pneumocystis carinii) is a fungus that is commonly found in the respiratory tract of many mammals, including humans (Wakefield, 2002). The fungus rarely causes problems in healthy individuals, but can cause problems in immunocompromised individuals – the most common manifestation being pneumocystis pneumonia. Common symptoms of this condition include nonproductive cough, shortness of breath, fever, and tachypnoea (rapid breathing).
Historically, pneumocystis pneumonia has been associated with patients receiving chemotherapy or undergoing organ transplantation, but more recently it has become a common opportunistic infection in patients with HIV/AIDS, causing significant morbidity and mortality (with an estimated mortality rate of 10–20% in this patient group during the initial infection). Surprisingly, the mortality rate amongst non-HIV patients is higher, at a reported 30–60% (Thomas and Limper, 2004). Thankfully, because of the introduction of HAART (Highly Active Anti-Retro viral Therapy), the incidence of pneumocystis pneumonia appears to be decreasing in HIV patients, but it still remains the most common opportunistic infection amongst these patients (Thomas and Limper, 2004).
Despite this positive progression, the risk of contracting pneumocystis pneumonia must be taken seriously and therefore it is suggested that the following patient groups should be offered chemoprophylaxis to minimise the risk of developing infection: patients with a CD4+ count (T-helper cell count) of less than 200 cells/μL, those who have received organ transplantation (e.g. lung transplant), those who have immune-deficiency diseases or severe protein malnutrition, and those who are undergoing chemotherapy (Green et al., 2007).
Co-trimoxazole is considered the agent of choice for the management of pneumocystis pneumonia, being used to treat active infection and to prevent infection in patients who are at risk (see above) (Grimwade and Swingler, 2003, 2006; Green et al., 2007). For treatment of mild and moderate disease, high-dose co-trimoxazole is given orally, but in cases where the disease is severe and a rapid response to treatment is needed, high-dose co-trimoxazole is given by intravenous infusion. When used as prophylaxis, co-trimoxazole is given at a lower dose than when it is given to treat active infection. Typically, for an adult, a dose of 960 mg daily is used for prophylaxis,6 but in cases where compliance is a problem (which may often be the case in developing countries), the same dose may be taken on alternate days without significantly affecting the efficacy (Green et al., 2007).
3.1.6.3 Nocardiosis
Nocardia are species of aerobic Gram positive bacteria commonly found in the soil, where they are involved in the decomposition of plant material. In some cases, however, certain species of Nocardia can be pathogenic to humans; for example, N. asteroides has been shown to cause pulmonary disease, while Nocardia brasiliensis can cause skin lesions (Saubolle and Sussland, 2003). These infections are most commonly associated with patients who have a compromised immune system, but can also occur in healthy patients who have a competent immune system – although this is less likely.
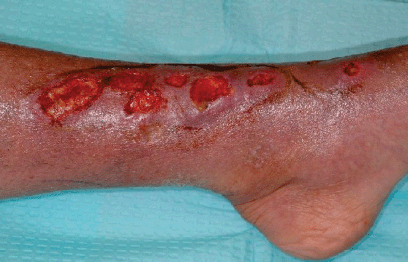
Nocardia can affect many organ systems, leading to different clinical forms of the disease. For example, pulmonary nocardiosis, the most common clinical form, is thought to be acquired via inhaling contaminated nocordial particles, while the introduction of Nocardia through the skin by a puncture wound or trauma (e.g. while gardening) can result in cutaneous nocardiosis (Figure 3.1.6). Other clinical forms of nocardiosis can include CNS infection (including brain abscesses), eye infection, and systemic (or disseminated) disease, which occurs when two or more body sites are affected.
Figure 3.1.6 A form of cutaneous nocardiosis caused by traumatic implantation of Nocardia brasiliensis in the foot (Reprinted from ‘Lymphocutaneous Nocardiosis Caused by Nocardia brasiliensis’, Dermatology Grand Round Cases, American Osteopathic College of Dermatology, Online, [http://www.aocd-grandrounds.org/case_46.shtml] 2006, with permission of J. Greg Brady.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


