Chapter 11
Stress and Disease
Margaret F. Clayton and Kathryn L. McCance

When thinking about stress, one must consider the factors producing a perception of stress. Stress begins with a stimulus that the brain perceives as stressful, which in turn promotes adaptational and survival-related physiologic responses. These responses can become dysregulated and cause pathophysiologic conditions.1 Another way to think about stress involves short- or long-term stressors and a discussion of the physiologic responses to these different types of events. Today most researchers feel that acute stress is considered to be immunoenhancing (protective) while chronic stress is now thought to be immunosuppressive (destructive).1
Historical Background and General Concepts
Walter B. Cannon used the term stress to encompass both physiologic and psychologic ideas as early as 1914.2 He applied the engineering concept of stress and strain in a physiologic context and believed that emotional stimuli also were capable of causing stress. In 1946, Hans Selye popularized these same findings, viewing stress as a biologic phenomenon.3 Originally, Selye inadvertently discovered the biologic syndrome of stress while he was attempting to discover a new sex hormone by injecting crude ovarian extracts into rats.3 He repeatedly found that three structural changes occurred: (1) enlargement of the cortex of the adrenal gland, (2) atrophy of the thymus gland and other lymphoid structures, and (3) development of bleeding ulcers in the stomach and duodenal lining. Selye soon discovered that these manifestations were not specific to injected ovarian extracts but also occurred after he exposed the rats to other noxious stimuli, such as cold, surgical injury, and restraint. He called these stimuli stressors. Selye concluded that this triad or syndrome of manifestations represented a nonspecific response to noxious stimuli, naming it the general adaptation syndrome (GAS). He identified three successive stages of the GAS: (1) the alarm stage or reaction, in which the central nervous system (CNS) is aroused and the body’s defenses are mobilized (e.g., “fight or flight”) (Figure 11-1); (2) the stage of resistance or adaptation, during which mobilization contributes to “fight or flight”; and (3) the stage of exhaustion, where continuous stress causes the progressive breakdown of compensatory mechanisms (acquired adaptations) and homeostasis. Exhaustion marks the onset of certain diseases (diseases of adaptation).
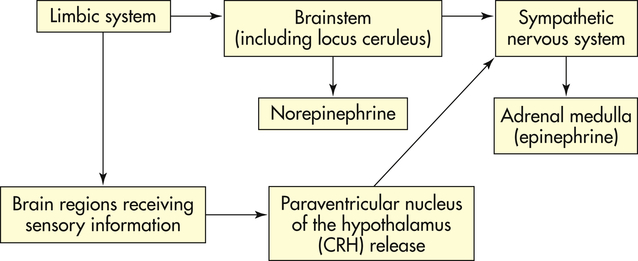
Initially one becomes alarmed by a stressor that activates the hypothalamus and sympathetic nervous system (see Figures 11-1 and 11-2). The resistance or adaptation phase begins with the actions of the hormones cortisol, norepinephrine, and epinephrine. Exhaustion (also known as allostatic overload; discussed later) occurs if stress continues and adaptation is not successful, ultimately causing impairment of the immune response, heart failure, and kidney failure, leading to death.
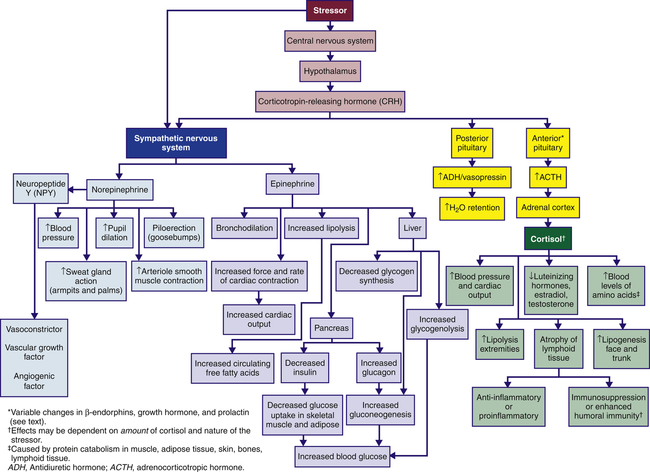
From a physiologic perspective, what is emerging across the disciplines involved—molecular biology, immunology, neurology, endocrinology, and behavioral science—is a more holistic and complex model that involves biochemical relationships of the CNS, autonomic nervous system (ANS), endocrine system, and immune system that cause the stress responses identified by Selye. More simply, these relationships are often cited together as the hypothalamic-pituitary-adrenal (HPA) axis (Figure 11-3). The reader may find it helpful to become familiar with the ANS function content in Chapter 15, p. 470.
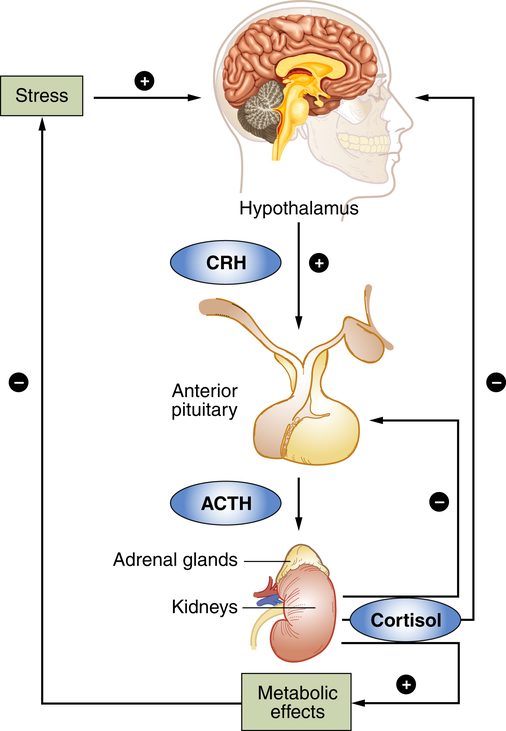
The response to stress begins in the brain. The hypothalamus is the control center in the brain for many hormones including corticotropin-releasing hormone (CRH). ACTH, Adrenocorticotropic hormone.
In sequence, the hypothalamus secretes corticotropin-releasing hormone (CRH), which binds to specific receptors on pituitary cells that, in turn, produce adrenocorticotropic hormone (ACTH). ACTH is then transported through the blood to the adrenal glands located on the top of the kidneys. After binding to specific receptors on the adrenal glands, the glucocorticoid hormones (primarily cortisol; from the adrenal cortex) are released. Cortisol initiates a series of metabolic changes discussed in the section Glucocorticoids: Cortisol; however, overall, these hormones are thought to enhance immunity during acute stress and suppress immunity during chronic stress because of prolonged exposure and increased concentration.1
Concepts of Stress
Selye believed that stressors cause a general or nonspecific response. However, research in the past 50 years has shown the remarkable sensitivity of the central nervous system and endocrine system to psychologic influences (emotion is included in psychologic and social stress and acts through psychologic mechanisms). Thus, although Selye’s identification of the GAS is regarded as tremendously important and the cornerstone of stress research, the idea that stress is a purely physiologic response is vastly oversimplified. In the mid-1950s, studies showed that activation of the adrenal cortex occurred in humans in response to psychologic stressors,4 in monkeys with conditioned emotional responses,5 and in humans subjected to a stressful interview technique.6 In the early 1960s, researchers found that plasma cortisol levels in groups of subjects increased while they watched war movies and decreased while they viewed Disney nature films.7,8 Mason later demonstrated that the initiation of the GAS depended on psychologic factors surrounding the stressors.9 He also showed that various factors, such as degrees of discomfort or unpleasantness or suddenness of the stress, could account for the presence or absence of physiologic stress responses.9
The term stress has been used persistently and widely across many disciplines despite numerous disagreements over its definition. Nevertheless, in recent years stress has been more usefully defined as a transactional or interactional concept. Transactionally, stress is viewed as the state of affairs arising when a person relates to (i.e., interacts or transacts with) situations in certain ways. People are not disturbed by situations per se but by the ways they appraise and react to situations. In general, a person experiences stress when a demand exceeds a person’s coping abilities, resulting in reactions such as disturbances of cognition, emotion, and behavior that can adversely affect well-being. Moreover, psychologic stressors can elicit reactive or anticipatory stress responses. The reactive response is a physiologic response derived from psychologic stressors. For example, the stress of an examination may produce an increased heart rate and dry mouth in the unprepared student. The anticipatory response occurs when physiologic responses develop in anticipation of disruption of the optimal steady-state, also known as homeostasis. These anticipatory responses can be generated either by species-specific innate programs, such as reacting to the presence of predators and unfamiliar situations, or by experience-dependent memory programs created by conditioning.10 Anticipatory responses are learned responses under fine control by regions located in the brain’s limbic system. These regions are those most frequently associated with learning and memory and include the hippocampus, amygdala, and prefrontal cortex. In order for these regions to elicit a stress response, the paraventricular nucleus (PVN) of the hypothalamus must be stimulated. The limbic structures rarely interact directly with the PVN and are believed to influence the stress response through intermediary neurons, some of which are primarily used for the reactive response.
Today there is evidence implicating stress as a precipitating factor for some diseases and conditions and as an exacerbating factor, worsening symptoms and outcomes, in others, such as atherosclerosis, irritable bowel syndrome, ulcers, asthma, autoimmune disorders, anxiety, delayed wound healing, chronic pain and fatigue syndromes, reproductive dysfunction, diabetes, and depression.9–11 In addition, evidence published since 2000 generally supports a relationship between stress and human immunodeficiency virus (HIV) progression.12
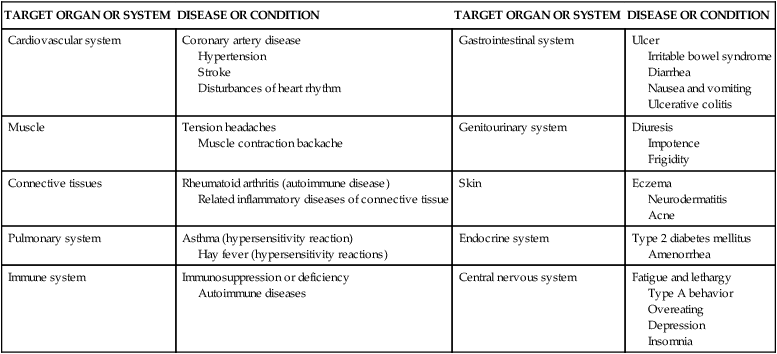
Psychologic stress may cause or exacerbate (worsen) several disease states, including many of the diseases implicated as the leading causes of death in the United States (Table 11-1). For example, stress-induced chronic inflammation is suggested as being important in the functional decline that leads to frailty, disability, and untimely death; cardiovascular disease; and infectious diseases.12–16 Additionally, important effects of acute emotional stress, such as the death of a loved one, on the heart muscle include three areas: (1) left ventricular contractile dysfunction, (2) myocardial ischemia, and (3) disturbances of heart rhythm (see What’s New? Acute Emotional Stress and Adverse Heart Effects). Other examples include research linking stress to the recurrence of genital herpes virus in women with HIV.13 As evidence has mounted concerning the important role that stress plays in certain disease processes, research has focused on the mechanisms responsible for these mind-body interactions. Along with a greater understanding of the relationship between the human stress response and disease, new strategies for treatment of stress-related disorders are emerging. It has recently been demonstrated that the interactions among social, psychologic, biologic, and behavioral factors are inherent in the causes and courses of many diseases. Molecular biologists, immunologists, neurologists, clinicians, and behavioral scientists are now exploring the role of the other half of the mind-body (dualistic) model—that is, the mind. What is emerging is a more holistic and complex model of health and disease states. This model involves the biochemical relationships of the central and autonomic nervous systems, the endocrine system, and the immune system and their relationships to stress-elicited coping behaviors, such as smoking and poor diet, that can also modify the integrity of the immune system. Discoveries of these complex links have led to the creation of the field of psychoneuroimmunology.
TABLE 11-1
EXAMPLES OF STRESS-RELATED DISEASES AND CONDITIONS
| TARGET ORGAN OR SYSTEM | DISEASE OR CONDITION | TARGET ORGAN OR SYSTEM | DISEASE OR CONDITION |
| Cardiovascular system | Coronary artery disease Hypertension Stroke Disturbances of heart rhythm | Gastrointestinal system | Ulcer Irritable bowel syndrome Diarrhea Nausea and vomiting Ulcerative colitis |
| Muscle | Tension headaches Muscle contraction backache | Genitourinary system | Diuresis Impotence Frigidity |
| Connective tissues | Rheumatoid arthritis (autoimmune disease) Related inflammatory diseases of connective tissue | Skin | Eczema Neurodermatitis Acne |
| Pulmonary system | Asthma (hypersensitivity reaction) Hay fever (hypersensitivity reactions) | Endocrine system | Type 2 diabetes mellitus Amenorrhea |
| Immune system | Immunosuppression or deficiency Autoimmune diseases | Central nervous system | Fatigue and lethargy Type A behavior Overeating Depression Insomnia |
Psychoneuroimmunologic Mediators of Stress
Psychoneuroimmunology (PNI) is the study of how the consciousness (psycho), brain and spinal cord (neuro), and the body’s defenses against infection and abnormal cell division (immunology) interact. Psychoneuroimmunology assumes that all immune-mediated diseases result from interrelationships among psychosocial, emotional, genetic, and behavioral factors with the neurologic, endocrine, and immune systems.11,17,18 The immune system is integrated with other physiologic processes and is sensitive to changes in CNS and endocrine functioning, such as those that accompany psychologic states. Stressors can elicit the stress response or stress system through the action of the nervous and endocrine systems. Stressors include infection, noise, decreased oxygen supply, pain, malnutrition, heat, cold, trauma, prolonged exertion, radiation, responses to life events (including anxiety, depression, anger, fear, loss, and excitement), obesity, old age, drugs, disease, surgery, and medical treatment.
Although the field of PNI is relatively new, and a body of scientific literature is present, the subject of PNI has caused strenuous scientific debate, especially with respect to the causal role of personality in cancer mortality and morbidity.19 For example, mouse models suggest a strong link between stress and breast cancer progression, yet this effect has not been consistently found in humans.19,20 What is certain, however, is that hormones released by the stress response influence many metabolic systems and corresponding physiologic events, even if the mechanisms and subsequent physiologic events are yet to be fully understood. Further, sufficient data now exist to conclude that immune modulation by psychosocial stressors or interventions leads directly to health outcomes.21–37 The strongest support to date for the link between psychosocial stressors and health outcomes remains in studies of infectious disease and wound healing.38–43 However, a very recent meta-analysis of 10 studies conducted among persons living in England found that any level of psychologic distress is associated with increased mortality and increased risk of death from cardiovascular disease, external causes, and cancer (albeit only at higher levels of distress); 68,000 persons were used in this study and personal factors, such as age, smoking, and alcohol use, were adjusted accordingly.44
Stress Response
The stress response is initiated by the central nervous system and endocrine system (see Figure 11-1). Specifically, corticotropin-releasing hormone (CRH) is released from the hypothalamus, and influences the sympathetic nervous system, the pituitary gland, and the adrenal gland (see Figure 11-2). CRH is also released peripherally at inflammatory sites called peripheral, or immune, CRH. The activation of these systems redirects adaptive energy to the CNS and stressed body sites.
Where the stress response begins depends on whether the stressor is perceived or real. Perceived stressors elicit an anticipatory response that usually begins in the limbic system of the brain, the area responsible for emotions and cognition. The limbic system indirectly elicits both an endocrine stress response by stimulating neural pathways responsible for receiving sensory information and a central stress response by directly stimulating the locus ceruleus (LC) to release norepinephrine (see Figure 11-1). Norepinephrine release promotes arousal, increased vigilance, increased anxiety, and other protective emotional responses. Real stressors elicit a reactive response that can begin either in the limbic system or in regions of the brain receiving specific sensory information (see Figure 11-1). This information is then relayed to the PVN. The PVN stimulates the LC and both central and endocrine stress responses.
Central Stress Response
Better understanding is emerging regarding the physiology involved in meeting the demands and challenges of daily life. Some refer to these challenges as “stressors,” and the chronically stressed person may use the term “stressed out.” The physiology is complex, involving both protection and damage. As previously mentioned, glucocorticoids from the adrenal cortex in response to ACTH from the pituitary gland (see Figure 11-3) comprise the major stress hormones along with the catecholamines epinephrine and norepinephrine. Other central hormones/mediators also play a role including the proinflammatory and anti-inflammatory cytokines that are regulated by glucocorticoids and catecholamines (Figure 11-4).21
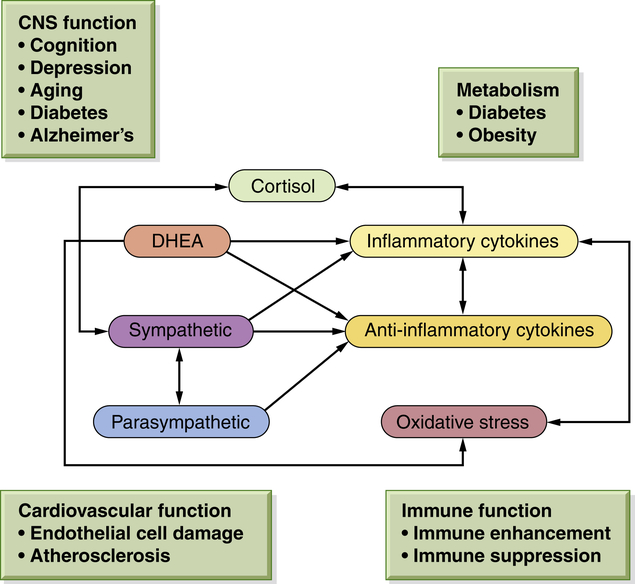
Nonlinearity means that when one mediator is increased or decreased, the subsequent compensatory changes in other mediators depend on time and level of change, causing multiple interacting variables. The inevitable consequences from adapting to daily life over time include changes in behavioral responses. For example, these changes include sleeping patterns, tobacco use, alcohol consumption, physical activity, and social interactions. These behavioral patterns are a part of the allostatic overload with chronic elevations in cortisol level, sympathetic activity, and proinflammatory cytokines, and a decrease in parasympathetic activity. (From McEwen BS: Eur J Pharmacol 583[2-3]:174–185, 2008.) DHEA, Dehydroepiandrosterone.
Catecholamines can increase proinflammatory cytokine production, causing, for example, increased heart rate and blood pressure. Glucocorticoids are known to inhibit this proinflammatory production; however, inhibition depends on dose and cell or tissue type.30 More simply, glucocorticoids can also promote inflammation depending on dose and cell type.31 The increased understanding of these effects suggests the possibility that chronic and dysfunctional HPA axis stimulation (as may occur during chronic inflammation) increases inflammation in the brain and other tissue, possibly contributing to other diseases including osteoporosis, metabolic disease (diabetes, obesity), and cardiovascular disease.32 Additionally, these interactions are nonlinear and are very complex.
Parasympathetic System
The parasympathetic system balances the sympathetic nervous system and, thus, also influences adaptation or maladaptation to stressful events. The parasympathetic system also has anti-inflammatory effects. The parasympathetic system opposes the sympathetic (catecholamine) responses, for example, by slowing the heart rate.31 Researchers evaluate the relative balance of the parasympathetic and sympathetic nervous systems using a technique known as heart rate variability (the measurement of R wave variability from heartbeat to heartbeat).
Allostasis is considered an adaptive physiologic response to stressful events (e.g., fight or flight).21 Chronic or dysregulated allostasis (long-term or chronic exaggerated responses to stress) can lead to disease. Allostatic load (Figure 11-5) is the individualized cumulative amounts of stressors that exist in our lives and that influence our physiologic responses. Allostatic load includes our genetic makeup, our lifestyle (including damaging health behaviors), daily events, and dramatic events (such as disasters).21,45 Over time this load exacts a toll on our bodies (i.e., “wear and tear”). Because the brain is a key player in deciding what is stressful, it is influential in determining when we have reached allostatic overload. Moreover, these responses are individualized in that what would be considered normal for one person is considered extremely stressful for another.45 Under conditions of allostatic overload, the parasympathetic system may decrease its restraint of the sympathetic system, resulting in increased or prolonged inflammatory responses.21,31 Physiologically, in response to acute and chronic stress some regions of the brain (the hippocampus, amygdala, and prefrontal cortex) may respond by undergoing structural remodeling, which can alter behavioral and physiologic responses (such as cognitive impairment or depression).21 Key mediators involved in allostatic overload (exaggerated pathophysiologic responses to stress) include the glucocorticoid cortisol, catecholamines (released from sympathetic nervous system activation), and proinflammatory cytokines; in addition, there is a decline in parasympathetic activity. A prevalent example is sleep deprivation resulting from excessive stress. Sleep deprivation has significant damaging effects including elevated evening cortisol level; elevated insulin and blood glucose levels; increased blood pressure; reduced parasympathetic activity; increased levels of proinflammatory cytokines; and increased concentrations of the gut hormone ghrelin, which increases appetite. Altogether, these alterations can lead to increased caloric intake, depressed mood, cognitive problems, and a host of other responses from insomnia.34
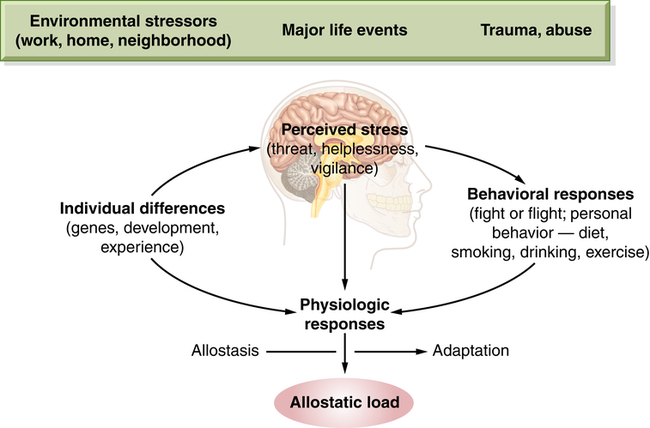
Stress processes arise from bidirectional communication patterns between the brain and other physiologic systems (autonomic, immune, neural, and endocrine). Importantly, these bidirectional mechanisms are protective, promoting short-term adaptation (allostasis). Chronic stress mechanisms, however, can lead to long-term dysregulation and promote behavioral responses and physiologic responses that lead to stress-induced disorders/diseases (allostatic load) that compromise health. (From McEwen BS: Eur J Pharmacol 583[2-3]:174–185, 2008.)
Neuroendocrine Regulation
The sympathetic nervous system (SNS) is aroused during the stress response and causes the medulla of the adrenal gland to release catecholamines (80% epinephrine and 20% norepinephrine) into the bloodstream. The adrenal medulla is actually an extension of the SNS because preganglionic fibers from the splanchnic nerve terminate in the medulla, where they innervate the chromaffin cells that produce the catecholamine hormones. Simultaneously, hypothalamic CRH stimulates the pituitary gland to release a variety of hormones, including antidiuretic hormone and oxytocin from the posterior pituitary gland, and prolactin, endorphins, growth hormone (GH), and adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. ACTH stimulates the cortex of the adrenal gland to release cortisol (see Figure 11-2).
Catecholamines
Circulating catecholamines essentially mimic direct sympathetic stimulation. Catecholamines cannot cross the blood-brain barrier and are synthesized locally in the brain. In response to stress, the chromaffin cells of the adrenal medulla produce large quantities of epinephrine and small amounts of norepinephrine. Once released, catecholamines circulate bound to the plasma protein albumin. The physiologic effects of the catecholamines on organs and tissues are summarized in Table 11-2. Norepinephrine regulates blood pressure by constricting smooth muscle in all blood vessels. During stress, norepinephrine raises blood pressure by constricting peripheral vessels; it dilates the pupils of the eye, causes piloerection, and increases sweat gland action in the armpits and palms (see Figure 11-2).
TABLE 11-2
PHYSIOLOGIC EFFECTS OF CATECHOLAMINES∗
| ORGAN/TISSUE | PROCESS OR RESULT |
| Brain | Increased blood flow; increased glucose metabolism |
| Cardiovascular system | Increased rate and force of contraction |
| Peripheral vasoconstriction | |
| Pulmonary system | Bronchodilation |
| Skeletal muscle | Increased glycogenolysis |
| Increased contraction | |
| Increased dilation of muscle vasculature | |
| Decreased glucose uptake and utilization (decreases insulin release) | |
| Liver | Increased glucose production |
| Increased glycogenolysis | |
| Adipose tissue | Increased lipolysis |
| Decreased glucose uptake | |
| Skin | Decreased blood flow |
| Gastrointestinal and genitourinary tracts | Decreased protein synthesis |
| Decreased smooth muscle contraction | |
| Increased renin release | |
| Increased gastrointestinal sphincter tone | |
| Lymphoid tissue | Acute and chronic stress inhibits several components of innate immunity, particularly decreasing number of natural killer cells |
| Macrophages | Inhibit and stimulate macrophage activity |
| Depend on availability of type 1/proinflammatory cytokines, presence or absence of antigenic stressors, and peripheral corticotropin-releasing hormone (CRH) |
∗Some of these responses require glucocorticoids (e.g., cortisol) for maximal activity (see text for explanation).
Data from Elenkov IJ, Chrousos GP: Ann N Y Acad Sci 966:290–303, 2002; Granner DK: Hormones of the adrenal medulla. In Murray RK et al, editors: Harper’s biochemistry, ed 25, New York, 2000.
Epinephrine is rapidly transported to and acts on several organs, but it is metabolized quickly, making it short-acting. Metabolically, epinephrine causes transient hyperglycemia (high blood glucose level), decreases glucose uptake in the muscles and other organs, and decreases insulin release from the pancreas. This is accomplished by activating enzymes whose actions promote glucose formation (gluconeogenesis) and glycogen breakdown (glycogenolysis) in the liver, while inhibiting glycogen formation. This prevents glucose from being taken up by peripheral tissue and preserves it for the CNS. Further, very little adrenal norepinephrine reaches distal tissue; thus, the effects caused by norepinephrine during the stress response are primarily elicited from the SNS.46,47
Table 11-2 summarizes other well-known effects of adrenal catecholamines. All of these effects prepare the body to take physical action: to fight or flee. Stressors commonly associated with catecholamine release by the adrenal medulla include exercise, thermal changes, and acute emotional states.
The catecholamines stimulate two major classes of receptors: α-adrenergic receptors and β-adrenergic receptors. These two classes are divided further into two subclasses: (1) α1 and α2 and (2) β1 and β2. Table 11-3 summarizes the actions of the two subclasses of adrenergic receptors. Epinephrine binds to and activates both α- and β-adrenergic receptors. Norepinephrine at physiologic concentrations binds primarily to α-adrenergic receptors.48
TABLE 11-3
PHYSIOLOGIC ACTIONS OF α- AND β-ADRENERGIC RECEPTORS
| RECEPTOR | PHYSIOLOGIC ACTIONS |
| α1 | Increased glycogenolysis; smooth muscle contraction (blood vessels, genitourinary tract) |
| α2 | Smooth muscle relaxation (gastrointestinal tract); smooth muscle contraction (some vascular beds); inhibition of lipolysis, renin release, platelet aggregation, and insulin secretion |
| β1 | Stimulation of lipolysis; myocardial contraction (increased rate, increased force of contraction) |
| β2 | Increased hepatic gluconeogenesis; increased hepatic glycogenolysis; increased muscle glycogenolysis; increased release of insulin, glucagon, and renin; smooth muscle relaxation (bronchi, blood vessels, genitourinary tract, gastrointestinal tract) |
Catecholamines can modify the numbers of cells of the immune system circulating in the blood.49 Injection of epinephrine into healthy human subjects is associated with a transient increase of the number of lymphocytes (e.g., T cells and natural killer [NK] cells) in the peripheral blood. Specifically, the levels of T cytotoxic and NK cells increase, whereas little change occurs in B lymphocytes. The main change involves the NK cells.49 Qualitatively, lymphocyte responsiveness of T and B lymphocytes is reduced. Similar quantitative and qualitative changes are found 5 to 6 minutes after exposure to a psychologic or physical stressor.50 However, the effects of acute elevation of catecholamine levels on the alteration of lymphocyte function are short-lived, lasting only about 2 hours.51
Glucocorticoids: Cortisol
The adrenal cortex is activated during stress by ACTH (see Figure 11-2), which increases adrenocortical secretion of glucocorticoid hormones, primarily cortisol (hydrocortisone is a synthetically produced but chemically identical version of cortisol). These steroid molecules reach all tissues, including the brain, easily penetrate cell membranes, and react with numerous intracellular glucocorticoid receptors. Because they spare almost no tissue or organ and influence a large proportion of the human genome, they exert significant diverse biologic actions.22 The feedback mechanisms of the HPA axis sense and determine the circulating glucocorticoid levels, whereas other tissues passively accept the actions of circulating glucocorticoids.22 Chronic dysregulation of the HPA axis, especially elevated cortisol level, has been linked to a wide variety of disorders including obesity, sleep deprivation, lipid abnormalities, hypertension, diabetes, atherosclerosis, loss of bone density, hippocampal atrophy, and cognitive impairment.45
Cortisol circulates in the plasma, both protein bound and free. The main plasma-binding protein is called transcortin or corticosteroid-binding globulin. The unbound, or free, fraction is approximately 8% of the total plasma cortisol and is biologically active.48 Cortisol mobilizes substances needed for cellular metabolism. One of the primary effects of cortisol is the stimulation of gluconeogenesis, or the formation of glucose from noncarbohydrate sources, such as amino acids or free fatty acids in the liver. In addition, cortisol enhances the elevation of blood glucose level promoted by other hormones, such as epinephrine, glucagon, and growth hormone. This action by cortisol is said to be permissive for the actions of other hormones. Cortisol also inhibits the uptake and oxidation of glucose by many body cells. The overall action of cortisol increases blood glucose concentration, thereby enabling the body to combat the stressor. The physiologic effects of cortisol are summarized in Table 11-4.
TABLE 11-4
PHYSIOLOGIC EFFECTS OF CORTISOL
| FUNCTIONS AFFECTED | PHYSIOLOGIC EFFECTS |
| Carbohydrate and lipid metabolism | Diminishes peripheral uptake and utilization of glucose; promotes gluconeogenesis in liver metabolism cells; enhances gluconeogenic response to other hormones; promotes lipolysis in adipose tissue |
| Protein metabolism | Increases protein synthesis in liver and decreases protein synthesis (including immunoglobulin synthesis) in muscle, lymphoid tissue, adipose tissue, skin, and bone; increases plasma level of amino acids; stimulates deamination in liver |
| Anti-inflammatory effects (systemic effects) | High levels of cortisol used in drug therapy suppress inflammatory response; inhibit proinflammatory activity of many growth factors and cytokines; however, over time some individuals may develop tolerance to glucocorticoids, causing an increased susceptibility to both inflammatory and autoimmune disease |
| Proinflammatory effects (possible local effects) | Cortisol levels released during stress response may increase proinflammatory effects |
| Lipid metabolism | Lipolysis in extremities and lipogenesis in face and trunk |
| Immune effects | Treatment levels of glucocorticoids are immunosuppressive; thus, they are valuable agents used in numerous diseases; the T-cell or innate immunity system is particularly affected by these larger doses of glucocorticoids with suppression of Th1 function or innate immunity; stress can cause a different pattern of immune response; these nontherapeutic levels can suppress innate (Th1) and increase adaptive (Th2) immunity—the so-called Th1 to Th2 shift; several factors influence this complex physiology and include long-term adaptations, reproductive hormones (i.e., overall, androgens suppress and estrogens stimulate immune responses), defects of hypothalamic-pituitary-adrenal axis, histamine-generated responses, and acute vs. chronic stress; thus stress seems to cause a Th2 shift systemically whereas locally, under certain conditions, it can induce proinflammatory activities and by these mechanisms may influence onset or course of infections and autoimmune/inflammatory, allergic, and neoplastic diseases |
| Digestive function | Promotes gastric secretion |
| Urinary function | Enhances excretion of calcium |
| Connective tissue function | Decreases proliferation of fibroblasts in connective tissue (thus delaying healing) |
| Muscle function | Maintains normal contractility and maximal work output for skeletal and cardiac muscle |
| Bone function | Decreases bone formation |
| Vascular system/myocardial function | Maintains normal blood pressure; permits increased responsiveness of arterioles to constrictive action of adrenergic stimulation; optimizes myocardial performance |
| Central nervous system function | Somehow modulates perceptual and emotional functioning; essential for normal arousal and initiation of daytime activity |
| Possible synergism with estrogen in pregnancy? | Suppresses maternal immune system to prevent rejection of fetus? |
Cortisol also has a powerful effect that reverses the insulin-induced suppression of hepatic gluconeogenesis, as well as basal levels of cortisol that stimulate the activity of hepatic enzymes responsible for glycogen and glucose production. The increased amino acid uptake into liver and the increased levels of glucose-producing enzymes favor the production of glucose. Although diseases of excess cortisol secretion, such as Cushing disease, produce characteristics of type 2 (non–insulin-dependent) diabetes mellitus, recent studies found that chronic stress also may facilitate the development of type 2 diabetes.52,53 Glucocorticoids contribute to the development of metabolic syndrome and the pathogenesis of obesity (Box 11-1). The mechanism for this action is under investigation, but it is believed that the development of diabetes is secondary to cortisol-induced obesity. Cortisol promotes lipogenesis in certain regions of the body and to a lesser extent promotes lipolysis in other regions by increasing the actions of lipolytic hormones, such as catecholamines and growth hormone. Chronic cortisol excess induces lipogenesis in the abdomen, trunk, and face, resulting in central obesity. Finally, glucocorticoids may affect fetal programming of the HPA axis by causing an adverse intrauterine environment because of alterations in cortisol secretion during pregnancy.54
BOX 11-1 GLUCOCORTICOIDS, INSULIN, INFLAMMATION, AND OBESITY

The induction of reactive oxygen species (ROS) generation and inflammation through the proinflammatory transcription factor, NF-kβ, activates most proinflammatory genes. Macronutrient intake, obesity, free fatty acids, infection, smoking, psychologic stress, and genetic factors increase the production of ROS. Interference with insulin signaling (insulin resistance) leads to hyperglycemia and proinflammatory changes. Proinflammatory changes increase the levels of TNF-α and IL-6, and also lead to the inhibition of insulin signaling and insulin resistance. Inflammation in pancreatic beta cells leads to beta-cell dysfunction, which in combination with insulin resistance leads to type 2 diabetes. CRP, C-reactive protein.
Data from Dallman MF et al: Endocrinology 145(6):2633–2638, 2004; Dandona P, Aljada A, Brandyopadhyay A: Trends Immunol 25(1):4–7, 2004; Kim SP et al: Diabetes 52:2453–2460, 2003; Masuzaki H et al: Science 94:2166–2170, 2001; Padgett DA, Glaser R: Trends Immunol 24(8):444–448, 2003; Spencer SJ, Tilbrook A: Stress 14(3), 2011.
Cortisol and the Immune System
Cortisol secretion during stress exerts beneficial effects by inhibiting initial inflammatory effects, for example, vasodilation and increased capillary permeability.32 Cortisol also promotes resolution and repair. These actions are mainly accomplished by facilitating the effects of glucocorticoid receptor (GR), namely, the transcription of genetic material (through DNA binding) within leukocytes.32 Because GR is so widely expressed, glucocorticoids influence virtually all immune cells. However, whether cortisol-induced effects are adaptive or destructive may depend on the intensity, type, and duration of the stressor; the tissue involved; and the subsequent concentration and length of cortisol exposure. Finally, glucocorticoids have been shown to induce T-cell apoptosis.32
The cytokines secreted by the Th2 cells act to inhibit Th1 cells and can promote adaptive immunity by stimulating the growth and activation of mast cells and eosinophils, as well as the differentiation of B-cell immunoglobulins. Thus, these cytokines are sometimes referred to as anti-inflammatory cytokines55 (Figure 11-6). Moreover, cytokines can act synergistically, antagonistically, or reciprocally. However, the roles of cytokines are highly complex and much remains unknown. Regardless, the decrease in Th1 activity and increase in Th2 activity is sometimes called a Th1 to Th2 shift. Individuals experiencing a Th1 to Th2 shift are more likely to experience allergic responses, infections, and temporary worsening of autoimmune conditions such as arthritis.
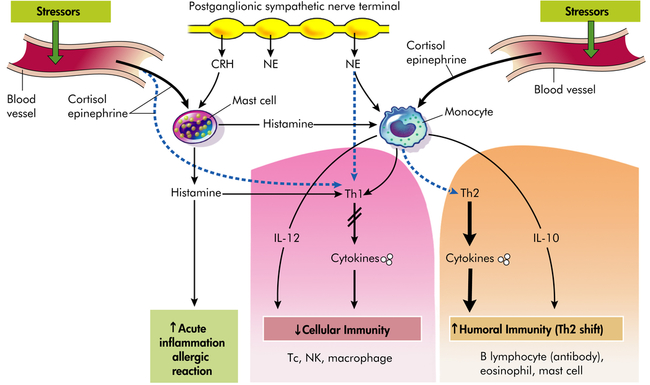
Adaptive immunity provides protection against multicellular parasites, extracellular bacteria, some viruses, soluble toxins, and allergens. Innate immunity provides protection against intracellular bacteria, fungi, protozoa, and several viruses. Type 1 cytokines or proinflammatory cytokines include IL-12, interferon-gamma (IFN-γ), and tumor necrosis factor-alpha (TNF-α). Type 2 cytokines or anti-inflammatory cytokines include IL-10 and IL-4. Solid lines (black) represent stimulation, whereas dashed lines (blue) represent inhibition (i.e., Th1 and Th2 are mutually inhibitory, IL-12 and IFN-γ inhibit Th2, and vice versa; IL-4 and IL-10 inhibit Th1 responses). Stress and CRH modulate inflammatory/immune and allergic responses by stimulating cortisol (glucocorticoid), catecholamines, and peripheral (immune) CRH secretion and by changing the production of regulatory cytokines and histamines. CRH (peripheral, immune), Corticotropin-releasing hormone; IL, interleukin; NE, norepinephrine; NK, natural killer cell; Tc, cytotoxic T cell; Th, helper T cell; dashed lines, decreased (inhibited); solid lines, increased (stimulation). (Redrawn from Elenkov IJ, Chrousos GP: Trends Endocrinol Metab 10[9]:359–368, 1999.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


