Statistical mechanics
When I took my first course in statistical mechanics from Hans Christian Andersen (yes, they are related) at Stanford, I said to myself, “…why haven’t I ever seen this before?” Well here it is – your introduction to stat mech. It is totally ridiculous to start a textbook in physical chemistry with statistical mechanics. It might just scare you off from the rest of the course. But let’s get something straight about this chapter. I neither expect nor want to you understand every detail of statistical mechanics – not even everything that is in this chapter. At least, not on your first reading of it. What I want you to learn is the structural basis for a molecular understanding of chemistry. What I really want you to understand is: (i) what is internal energy; (ii) what is a Maxwell–Boltzmann distribution; (iii) how entropy is related to configurations and is nicely presented in the Boltzmann formula and Sackur–Tetrode equation; (iv) what is a partition function; and (v) how does this all relate to the heat capacity? There is a lot of mathematics in between these points, but don’t let it get in the way. I don’t really intend to go into it in detail. However, I do not think it is quite viable in a serious textbook to write
- There is a thing called the molecule.
- A miracle happens, that is, statistical mechanics.
- Here are the equations for entropy, energy distribution, and heat capacity.
In the words of Gary Larson, “I think you should be more explicit here in step two.” So I have been. After you have completed your first course in physical chemistry, or before you enter graduate school, come back to this chapter and really try to understand the steps in between. Then go on to learn more. For now, concentrate most on the big picture and you will have a much easier time understanding the rest of physical chemistry.
Here for review and clarity, let us introduce a number of terms and definitions:
- System – the material in the process under study.
- Surroundings – the rest of the universe.
- Open system – system can exchange material with surroundings.
- Closed system – no exchange of material.
- Isolated system – no exchange of material or energy.
- Thermodynamic equilibrium – p, T, composition are constant and the system is in its most stable state. Also called chemical equilibrium.
- Thermal equilibrium – two systems have the same temperature.
- Metastable state – a system in which p, T and composition are constant; however, the system could relax to a lower energy state if it were sufficiently perturbed. A system in a metastable state is in a state of metastable equilibrium, and so can be described consistently by thermodynamic methods.
- Steady state – p, T and composition are constant but are maintained at these values in an open system by a balance of inward and outward fluxes of matter and energy.
- Isothermal – ΔT = 0, isobaric – Δp = 0, isochoric – ΔV = 0.
- Adiabatic in a thermodynamic sense means that the system exchanges no heat, q = 0.
- Diathermal barrier – allows for the transport of heat but not matter between systems.
- Temperature T in kelvin (absolute temperature) is related to temperature t in degrees Celsius by:

6.1 The initial state of the universe
We start at the beginning – or shortly after the beginning. Immediately after the Big Bang, the universe was in a rather uniform state in which the density of energy exceeded the density of matter. The rather (though not completely) uniform density corresponded to a much more ordered state of the universe than the one we observe now: an extremely inhomogeneous clumping of atoms into molecules and compounds and planets and stars (as well as plants and animals), and stars into galaxies, and galaxies into galactic clusters. From this highly ordered initial state sprang the natural tendency for systems to develop from whatever state they are in to less-ordered states. This is a tendency, not an irrefutable law, and work can be done to overcome this tendency. Experimentalists can use lasers to create initial states that will develop and return to their initial ordered state. But the recurrence time in these so-called ‘spin-echo’ experiments is extremely short and the number of atoms that will cooperate decays rapidly with time. Thus, even with the most care and best lasers, such an exceptional state of affairs can only be maintained for a fraction of a second.
In order to understand the tendency of isolated systems to equilibrate – to understand how systems evolve chemically – we need to understand the physics that controls and describes the interactions between atomic and subatomic particles. But even this is not enough. We need to understand that the material world around us is composed of microscopic atoms and molecules. Further, there are an enormous number of these molecules. A mere 10 ml of liquid water contains on the order of 1023 water molecules. Their microscopic nature (in particular their minute masses and distances between them) means that they do not always play by the rules of the macroscopic world. We will discuss much more about that when we treat quantum mechanics, but for now we concentrate on the effect of the enormous number of molecules.
The sheer number of molecules in a macroscopic sample leads to emergent behavior. The equations that describe the dynamics of individual atoms and molecules are symmetric in time and space. Nonetheless, we know that samples made up of atoms and molecules irreversibly evolve toward equilibrium. In addition, even if we have a complete description of the properties and dynamics of one molecule, there is no way for us to describe the phase transitions that a collection of these molecules undergoes. Both, the irreversible approach to equilibrium and phase transitions are examples of emergent behavior: behavior that cannot be predicted from the properties of one molecule but that emerges from the interactions of collections of molecules.
6.2 Microstates and macrostates of molecules
In order to derive the macroscopic behavior of matter from the microscopic properties of molecules, we need to derive the principles of statistical mechanics. Statistical mechanics starts with the conception of matter as being composed of N particles occupying volume V. This is not how thermodynamics was founded but, as we shall see, it allows us to derive the principles of thermodynamics on a molecular basis. The thermodynamic state of these N particles is called a macrostate. The macrostate is a description of the system in terms of collective thermodynamic parameters. We choose our macrostate to be isolated, that is, its internal energy U is held constant, as is N. To specify our system fully, we need only identify its composition in addition to N, V, and U. Should it be more convenient, we can use the methods of thermodynamics to specify our system in terms of other variables, such as N, the pressure p, temperature T, and composition. We will develop these thermodynamic methods in subsequent chapters.
If we want to know details of the individual particles – let us call them molecules though they may be molecules or atoms – then we need to define the microstate of the system. The microstate of a system is the description of the system in terms of the characteristics of each individual molecule. Each of the N particles is described quantum mechanically by a wavefunction ψk, and the total wavefunction of the system ψ contains a product of all the molecular wavefunctions ψ1 ψ2 …ψN (it may also contain some other terms to correctly account for the statistics of the system, as we shall see below). At this point we do not need to define what the wavefunction is mathematically. Each individual wavefunction is simply a mathematical function that describes a molecule.
At any finite temperature the molecules are constantly changing their states. The molecules collide, and these collisions change the velocities as well as the rotational and vibrational states of the molecules. Nonetheless, the number of molecules, their internal energy, as well as the volume remain constant. Thus, there is a large number Ω of microstates that correspond to the macrostate defined by N, U, and V. The fundamental postulate of statistical mechanics is the Gibbs postulate:
All possible microstates of an isolated assembly are equally probably.
Being a postulate, there is no general proof of this statement, though proofs for specific cases exist. Support of this postulate is derived from the vast number of correct results derived from it.
To describe our system more specifically, we need to know whether the particles are distinguishable or indistinguishable. This makes a difference in the statistics of the system. A one-component gas contains molecules that are identical and indistinguishable. All molecules are able to explore the entirety of the container, and there is no way to label individual molecules. Molecules arranged in a solid lattice are also identical, but because each is fixed to a lattice site, they can be labeled and distinguished.
We allow the N particles to equilibrate in a fixed volume V. In order to equilibrate, the molecules must interact; however, these interactions are not so strong as to interfere with identifying the molecules as individual particles. No reactions occur and for now we ignore any intermolecular interaction energy (essentially we will think in terms of ideal gases rather than real gases). Each of the N particles has an energy ϵj. Anticipating the results of quantum mechanics, we say that there is a level (state) that corresponds to each allowed value of energy and that each molecule occupies one of these levels. The total occupancy of level j is called the population nj. Another term for population is the occupation number. The total number of particles N is given by the sum of energy level populations over all allowed states

and the total internal energy is the sum of the energies of all molecules
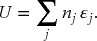
For any collection of N particles there are different ways for the nj particles to populate the ϵj energy states. That is, there are numerous microstates described by the different energy arrangements. An example is shown in Fig. 6.1. The number of ways that N distinguishable molecules can be distributed into j energy levels is given by:


Figure 6.1 A collection of seven objects (i.e., N = 7) is distributed over two different sets of states. The red set of states is nondegenerate; that is, there is only one state at each energy (the degeneracy g = 1). In the blue set, the degeneracy increases with increasing magnitude of j as gj = 2j + 1. Suggestively in this example, only two objects are allowed in each sublevel within each state. In the red set, the population of the j = 0 state is n0 = 4. In the blue set, the population of the j = 0 state is n0 = 2.
The total number of microstates Ω is the sum of t(n) for all allowed sets of populations that have a total internal energy U according to

The sum is over all distributions satisfying the constraint on internal energy. The total number of microstates is sometimes referred to as the weight.
What types of distribution satisfy the internal energy constraint? Consider a system with N = 3 and levels with energies ϵ0 = 0, ϵ1 = ϵ, ϵ2 = 2ϵ, ϵ3 = 3ϵ, …. If the total energy is U = 3ϵ, then we could put all of the particles in energy level ϵ1. Or we could put one particle in ϵ0, which requires that the other two go into ϵ1 and ϵ2, such that the sum of the energies 0 + ϵ + 2ϵ = 3ϵ. Or we could put two particles in ϵ0, which requires that the third occupy ϵ3. If the particles were indistinguishable, these would be the three allowed distributions. However, if the three particles are distinguishable – that is, if we have particles that are identifiable as A, B and C – then we also have to keep track of which particle has which energy. In this case, all of the distributions listed in Table 6.1 are possible.
Table 6.1 Allowed distributions for three distinguishable particles A, B, and C with U = 3ϵ and energy level structure ϵ0 = 0, ϵ1 = ϵ, ϵ2 = 2ϵ, ϵ3 = 3ϵ, ….
| A | ϵ | 2ϵ | 2ϵ | ϵ | ϵ | 0 | 0 | 3ϵ | 0 | 0 |
| B | ϵ | ϵ | 0 | 2ϵ | 0 | 2ϵ | ϵ | 0 | 3ϵ | 0 |
| C | ϵ | 0 | ϵ | 0 | 2ϵ | ϵ | 2ϵ | 0 | 0 | 3ϵ |
| n0 | 0 | 1 | 2 | |||||||
| n1 | 3 | 1 | 0 | |||||||
| n2 | 0 | 1 | 0 | |||||||
| n3 | 0 | 0 | 1 | |||||||
| t(n) | 1 | 6 | 3 | |||||||
There are a total of ∑t(n) = 10 microstates in Table 6.1. That is, there are 10 distinct ways to write the populations of the energy levels. We also see that there are three different classes of distributions (call them classes I, II, and III), but that each class has a certain number of permutations of how it can be written. Since each individual microstate is equally probable, the class with six different permutations is the most likely of the three classes to be present. Let us call this maximum value tmax. In this example, there is a 60% chance of finding the macrostate with U = 3ϵ in one of the microstates of class II. As N increases, the concentration of probability into the class that corresponds to tmax increases. For macroscopic quantities of molecules N is of the order of the Avogadro constant (1023 or so). For such astronomically high values of N, to a very good approximation, we can simply take

Therefore, we can rewrite Eq. (6.5) as

where the factorial in the denominator must be for the distribution that maximized t(n). Maximization is performed by using the method of undetermined multipliers and Stirling’s theorem. The method of undetermined multipliers, also called Lagrange multipliers, can be found in Appendix 1. From it we obtain the useful expression

Stirling’s theorem is used over and over again in statistical mechanics. It is derived in Appendix 2, and allows us to approximate the value of the logarithm of a very large number according to
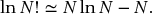
With these methods we find

We will determine the values of α and β later. The distribution described by Eq. (6.10) is the Boltzmann distribution. It describes the most probable constellation of the macrostate, which is the equilibrium state. This distribution is a set of population values – a set of values of nj – for which the value of Ω is a maximum. Other distributions are possible. These represent fluctuations away from the equilibrium distribution. In the example above, classes I and III represent fluctuations away from the equilibrium distribution of class II. As shown in Fig. 6.2, these fluctuations become increasingly less likely or, at least, the values of the fluctuations become increasingly small compared to the mean values of thermodynamic parameters as the number of particles approaches 1023 (a mole of atoms). However, fluctuations are substantial for particles on the nanoscale (<100 nm), and this is one reason why the behavior of nanoparticles can differ from our expectations of macroscopic samples.

Figure 6.2 The relative probability of a fluctuation (assuming Poisson statistics) is plotted as a function of the number of atoms, N. In the inset, the diameter of a spherical particle with the density of solid Si at 298 K is plotted along the right-hand axis as a function of number of atoms. Fluctuations on the order of 0.1% are to be expected for 40 nm-diameter particles, and this probability increases rapidly as the number of atoms decreases.
Note a subtle difference between the definitions of distribution and microstate. The microstate identifies the state of each molecule. The distribution specifies how many molecules (regardless of their identity) populate each of these microstates.
6.2.1 Directed practice
Chart the allowed distributions for four distinguishable particles A, B, C, and D with U = 3ϵ and energy level structure ϵ0 = 0, ϵ1 = 1, ϵ2 = 2, ϵ3 = 3, …. Calculate ∑t(n) and tmax.
6.3 The connection of entropy to microstates
In 1872, at a time when the very existence of molecules was still very much in doubt, Boltzmann related the entropy S of a gas to the probability of a gas being in a particular microstate. He did so in terms of the distribution function of molecular velocities f(vx, vy, vz). Planck’s quantum hypothesis – the idea that the energy states of matter are discreet (quantized) levels rather than continuous distributions – allowed Planck to generalize Boltzmann’s results to all thermodynamic systems.
From a thermodynamic viewpoint (as we shall define later), equilibrium in an isolated system is obtained when the entropy is a maximum. Statistical mechanics defines the equilibrium state as the most probable macrostate, the macrostate that corresponds to the maximum number of microstates. Therefore, the link between thermodynamics and statistical mechanics must lie in an equation that links the entropy S to the number of microstates Ω. The link, as derived by Boltzmann and Planck, is the exceedingly compact and important Boltzmann formula:

This equation is so fundamental and so groundbreaking that it is literally etched in stone – it appears on Boltzmann’s tombstone. The constant kB is the Boltzmann constant.
An a posteriori derivation of Eq. (6.11) is amazingly simple, but only because we know the answer. The deep insight of the work of Boltzmann and Planck should not be underestimated. We start with the idea that there is a ground energy state, a perfect crystal in internal equilibrium. At T = 0, all of the particles must enter this lowest energy state. Thus, each molecule has the same energy and there is only one microstate that describes the system – that is, Ω = 1. This single state is a state of perfect order for which, according to the third law of thermodynamics, S = 0. Note that ln 1 = 0, suggesting but not proving a logarithmic relationship

S is an extensive property. It depends on how much of a system we have. Take two independent assemblies with entropies S1 and S2 and corresponding numbers of microstates Ω1 and Ω2. The total entropy S of the two systems is given by the sum

The total number of microstates of the combined system Ω is obtained by pairing each of the Ω1 microstates of system 1 with each of the Ω2 microstates of system 2. The value of Ω is given by the product

Using the Ansatz that Eq. (6.11) is satisfied by simply multiplying ln Ω by a constant, which we call kB, we can show that Eqs (6.13) and (6.14) are confirmed. That is, assuming that Eq. (6.11) holds, then on substitution from Eq. (6.14)

we arrive at the result of Eq. (6.13). We will leave it to mathematicians to prove uniqueness. That is, not only are Eqs (6.11), (6.13) and (6.14) consistent, but also the only expression that makes Eqs (6.13) and (6.14) consistent is Eq. (6.11). If the statistical mechanical definition of entropy had been developed first, kB could have been assigned any arbitrary value. However, to ensure consistency with thermodynamics, its value is set by derivation of the ideal gas law, which we do below, and to satisfy an equation derived from the study of heat, which is shown in Chapter 9 to be a mathematical expression of the second law of thermodynamics,

6.3.1 Example
Calculate the entropy change when a mole of CO molecules in a perfect crystal at T = 0 K is transformed into an ensemble in which the orientation of the CO dipoles flip randomly between parallel and antiparallel alignments.
There are NA = 6.02 × 1023 molecules in a mole. A perfect crystal at 0 K has but one microstate, Ω = 1. The randomness introduced by the two orientations of the CO dipole introduces on the order of  microstates. Thus, the entropy change is
microstates. Thus, the entropy change is


Note that this value of ΔS is the value for a sample with 1 mole of molecules, the value of the molar entropy change is ΔSm = 5.76 J K−1 mol−1. This extra entropy that is present due to disorder even at 0 K is known as residual entropy.
6.4 The constant α: Introducing the partition function
The population of level j is given by

The exponential term involving β is called the Boltzmann factor. The total number of molecules is given by the sum of the populations, thus,

The exponential term in the summation is sufficiently important that we give it the name partition function and the symbol Z,

The partition function tells us how the population is partitioned into the various levels in accord with their respective Boltzmann factors. Solving for eα and substituting from Eq. (6.19) yields,

Thus, the populations of the individual levels are given by

The value of α is, therefore,

Its meaning is related to the chemical potential, which we will define later.
6.4.1 The value of β
Take a system with constant N and V. Now change the internal energy by adding an infinitesimal amount of heat dq. No pV work is done and the change in internal energy is related to the added heat by

The energy level structure does not change at constant volume as shown for example in Eq. (6.57) below. Therefore, dϵj = 0 and the heat transfer is related to a change in populations according to

The system will equilibrate by maximizing Ω, which will change according to

Using an expression found during the derivation of the Boltzmann distribution, Eq. (6.8), we substitute for dln Ω/dnj, which gives

However at constant N,  and then upon substitution from Eq. (6.24)
and then upon substitution from Eq. (6.24)

Solving for β and substituting from the second law of thermodynamics Eq. (6.16) dq = T dS,

Then from Eq. (6.11)

Proof that the value of kB truly is what is known as the Boltzmann constant is actually given unambiguously by the derivation of the equation of state of the ideal gas, which we will perform below.
6.5 Using the partition function to derive thermodynamic functions
For any one arrangement of N identical distinguishable molecules (such as molecules localized at lattice points), the partition function is

This assumes that the energy levels ϵj are nondegenerate. We will deal with degeneracy (more than one level at the same energy) in a moment. The localized particles can be arranged in many different ways. For the assembly as a whole, the partition function is

where the values of EN include all possible values of the total energy of the assembly. EN is the sum of all values of ϵ and the summation is over all possible values of EN.
Consider two distinguishable particles A and B, in which case


and

Thus

However, since A and B are identical ZA = ZB = Z and ZA ZB = Z2. This argument is easily generalized to N particles, for which

Until now we have considered the energy levels to be nondegenerate; that is, there is one and only one level at each value of energy. However, it is possible for more than one wavefunction – the quantum mechanical description of an energy level – to have the same energy. If more than one level has the same energy, then we say that the levels are degenerate. Another term for degeneracy is statistical weight, and this gives you an idea of how to account for degeneracy should it occur. If the number of levels at the same energy is gj, we say the degeneracy of the jth level is gj and we need to count each degenerate level gj times.
Including the effects of degeneracy, we can now summarize the results we have derived so far.

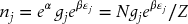





To these results derived from statistical mechanics we can now add the following results from thermodynamics, which will allow us to derive all thermodynamic functions directly from statistical (molecular) arguments. These thermodynamic results will be derived in Chapters 7–11.
Helmholtz energy

Gibbs energy

Enthalpy

Entropy

Pressure

Temperature

6.5.1 Example
Take the derivative of the Helmholtz energy at constant volume with respect to temperature and derive an equation that relates it to the entropy.
Taking the derivative of Eq. (6.44), we obtain

The definition of the heat capacity at constant volume is

therefore,

From the second law of thermodynamics, Eq. (6.16), we know1 that

The heat exchanged along a reversible path at constant volume is dqV, rev = CV dT. Upon substitution into Eq. (6.53), we obtain

Therefore

For purists, having specified the path as reversible and isochoric, we can equate dS/dT with (∂S/∂T)V. Substituting for CV in Eq. (6.52), yields the desired expression.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


