The number of tissue blocks varies depending on the size of the spleen, macroscopic findings, and the clinical setting. Generally, I submit from 4 to 10 blocks for histologic studies. The choice of fixative is optional; the only requirement is sufficiently thin tissue blocks and time for adequate fixation. In practice, more than half the cassettes are placed in formalin and the remainder in B5 solution. After approximately 30 minutes to 1 hour, the blocks should be trimmed if adequately thin sections were not initially obtained. At this time, the slowness of the penetration of the fixative into the splenic parenchyma, probably because of blood within the tissues, becomes obvious. Overnight fixation is recommended; however, depending on when the specimen was received and the urgency of the case, pilot sections may be sent for processing the same night.
If the spleen harbors a lymphoid neoplasm, the guidelines proposed by the Association of Directors of Anatomic and Surgical Pathology for the splenic pathology report are recommended, including the employment of the World Health Organization (WHO) classification (13,14).
DISORDERS PREDOMINATING IN THE SPLENIC WHITE PULP
In the histologic examination of a spleen, the surgical pathologist should determine on low magnification whether the pathologic process primarily involves the white or red pulp or whether the architecture appears intact. For those disorders disposed to affect mainly the white pulp, expansion of the white pulp is observed. The expanded white pulp will appear as a nodule or series of nodules against the diffuse red pulp background. The term nodular is employed simply as a description in this context and does not imply a follicular lymphoma (FL). The scope and pattern of nodular expansion is diverse, depending on the etiology and the extent of the disease. The major disorders leading to an enlarged white pulp are analogous to the disorders leading to lymphadenopathy. Essentially, white pulp expansion is either the result of lymphoid hyperplasia or a malignant lymphoproliferative disorder (Table 18.1).
REACTIVE FOLLICULAR HYPERPLASIA
Secondary germinal centers within the splenic white pulp are normally found in children, but they are less common in adults, in whom reactive follicles imply antigenic stimulation. Reactive follicular hyperplasia of the white pulp is the predominant morphologic abnormality in spleens that is associated with blood cytopenia and hypersplenism. The cytopenia is a reduction in all major hematopoietic lines or a decrease in one specific hematopoietic element, such as the diminished neutrophils found in Felty syndrome. Reactive follicular hyperplasia also may be the major pathologic abnormality in spleens from patients with hypersplenism in whom the hypersplenic state does not precisely correspond to a well-defined clinical condition and in whom the cause of the antigenic stimulus is unknown. Such spleens may be dramatically enlarged, and the splenic cut surface may contain multiple pale-tan nodules indistinguishable macroscopically from lymphoma (Fig. 18.2). Spleens with idiopathic reactive follicular hyperplasia and Felty syndrome may weigh more than 1000 g (5,15).
The reactive follicles in the splenic white pulp are similar to reactive follicles in lymph nodes in that they often vary in both size and shape. This observation, coupled with a mixed follicular center cell population and tingible body macrophages, serves to distinguish reactive follicular hyperplasia of the spleen from FL. At times, reactive follicles may be impossible to distinguish from the follicles of early-stage grade 2 FL, particularly if fixation is suboptimal; immunologic studies may be necessary for an accurate diagnosis.
Reactive follicular hyperplasia, however, is rarely an isolated pathologic finding. Unlike the situation in lymphomas, the antigenically stimulated white pulp usually is accompanied by a proliferation of mature lymphocytes and plasma cells in the red pulp as a probable manifestation of antibody production against a specific hematopoietic element (1,15). Light microscopic evidence of granulocytic phagocytosis is not apparent in spleens removed from patients with Felty syndrome; however, the red pulp sinuses and cords are expanded with increased numbers of macrophages (16). In some patients with autoimmune disorders such as systemic lupus erythematosus and even rheumatoid arthritis, the spleen may exhibit focal to extensive histiocytic necrosis in the white pulp with apoptosis and karyorrhectic debris but without hematoxylin bodies (17); associated rare Epstein-Barr virus (EBV)–positive cells as demonstrated by in situ hybridization for EBV-encoded RNA (EBER) often are present.
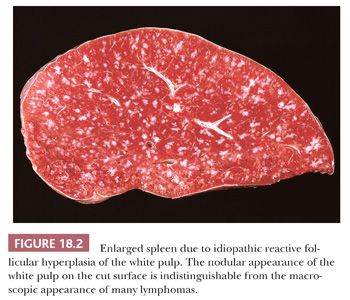
In immune thrombocytopenia (ITP), foamy macrophages or ceroid-laden histiocytes may be observed within the red pulp cords, attributable to the increased phagocytosis of platelets (Fig. 18.3) (18,19). The more numerous ceroid histiocytes within the red pulp are most likely related to a relative sphingomyelinase deficiency (20). Scanning electron microscopy of spleens removed from patients with ITP reveals changes in the microcirculatory pathways that consist of increased vascularization of the white pulp and marginal zones and absence of the marginal sinuses (21). These microcirculatory changes likely increase the exposure of platelets to splenic macrophages and add to increased platelet destruction in ITP. Splenectomy is effective in most patients with ITP because this procedure impedes the clearance of autoantibody-coated platelets by receptors on splenic macrophages, as well as impeding the interactions between T and B cells that are involved in the synthesis of antiplatelet and antimegakaryocytic antibodies (22).
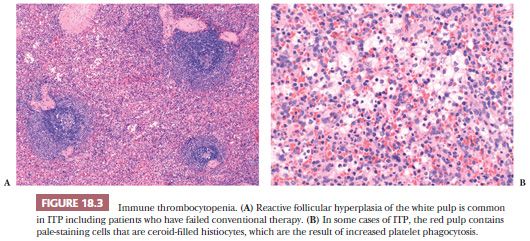
Although reactive follicular hyperplasia of the splenic white pulp and plasmacytosis with increased B cells in the red pulp, together with some degree of ceroid histiocytosis and even extramedullary hematopoiesis, are the recognized features of ITP, not all of these findings are apparent in many cases (23,24). Reactive follicular hyperplasia may be minimal to nonexistent despite a well-established clinical diagnosis and usually is a consequence of prior steroid therapy (25). Nonetheless, in steroid-failed ITP splenectomy specimens, aggregates of proliferating lymphocytes within white pulp that are distinct from germinal centers are found and are referred to as proliferative lymphoid nodules (26); such nodules are postulated to be the sites of autoantigen stimulation in ITP. Platelet phagocytosis endures in ITP spleens following steroid therapy and may be evident in touch imprints and in cordal macrophages as a series of basophilic clumps or granules (Fig. 18.4) (11,25). For ITP patients treated with rituximab and who do not respond, splenic B cells are significantly reduced, but long-lived plasma cells persist and activated CD8-positive T cells increase to imply their involvement in platelet destruction (27). When the constellation of splenic morphologic findings is not present in a setting of clinically established ITP following systemic therapy, a descriptive diagnosis is advised. A description may be especially relevant in ITP spleens that are morcellated in the course of a laparoscopic splenectomy (28).
Secondary germinal centers and B-cell hyperplasia of the white pulp also are commonly prominent in thrombotic thrombocytopenic purpura (TTP) (29). The reactive follicles often are accompanied by periarteriolar concentric fibrosis, which suggests an immunologic role in this condition. In TTP, arterial thrombi and hyaline subendothelial deposits of platelet and platelet-related material are the most characteristic pathologic alterations. In this context, hemophagocytosis, iron deposition in histiocytes, and extramedullary hematopoiesis are other common findings in TTP spleens.
Other causes of splenic reactive follicular hyperplasia include the rare cases of systemic Castleman disease, the acquired immunodeficiency syndrome (AIDS), and an unusual localized tumorous form (30–32). In systemic Castleman disease, the spleen exhibits changes of the hyaline vascular and plasma cell type similar to the changes found in Castleman disease involving lymph nodes (30). In some cases of nonspecific florid reactive follicular hyperplasia of the spleen, isolated white pulp segments also may have hyaline vascular changes indistinguishable from those seen in systemic Castleman disease.
Most descriptions of the spleen from patients with AIDS are derived from postmortem studies in which the spleen itself is characterized frequently by lymphoid depletion, with atrophy of the marginal zone, signs of opportunistic infections, and often deposition of perivascular para-amyloid material (31,33,34). Nonetheless, splenic AIDS cases may exhibit florid lymphoid hyperplasia in which plasmacytosis and immunoblastic proliferation affecting the white and red pulp are also seen (35). Some cases of ITP may be related to the prodromal phase of AIDS; the spleens from these cases exhibit reactive follicular hyperplasia (36). Moreover, the degree of reactive follicular hyperplasia is more prominent and splenic weights are greater in comparison to patients with ITP who do not have AIDS (37).
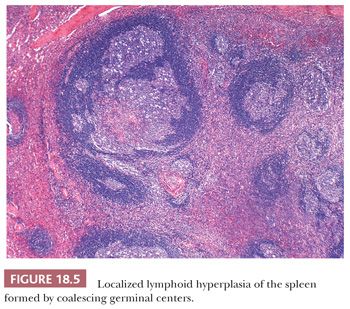
The localized form of reactive lymphoid hyperplasia is characterized by isolated tumor-type nodules on the splenic cut surface (32). Although the macroscopic appearance suggests lymphoma, particularly because the majority of cases originally were derived from spleens obtained at staging laparotomy for lymphoma, microscopic studies indicate that the nodules mainly are a result of focal coalescence of white pulp germinal centers (Fig. 18.5). The etiology is unknown, but the formation of a localized tumorous area of splenic lymphoid hyperplasia is thought to be analogous to florid lymphoid hyperplasia confined to a solitary enlarged lymph node (32).
REACTIVE NONFOLLICULAR HYPERPLASIA
Reactive lymphoid hyperplasia of the splenic white pulp in which secondary germinal centers are absent is analogous to diffuse lymphoid hyperplasia found in lymph nodes. The hyperplastic white pulp may be composed mainly of small lymphocytes, but, most frequently, a range of lymphoid transformation that includes plasma cells and immunoblasts is discovered. This form of hyperplasia may be observed in acute infections, such as viral infections like AIDS, and graft rejection, but, frequently, the etiology is unknown (1).
The most blatant examples of reactive nonfollicular lymphoid hyperplasia that I have observed occurred in examples of acute ITP (Fig. 18.6) (5). These cases were associated with florid immunoblastic proliferation involving both the white and red pulp in the presence of immunoblastic infiltration of the subintima of trabecular veins and the splenic capsule. The immunoblastic proliferation resembled an acute viral infection, such as infectious mononucleosis (38), but a viral infection, including infectious mononucleosis, could not be demonstrated.
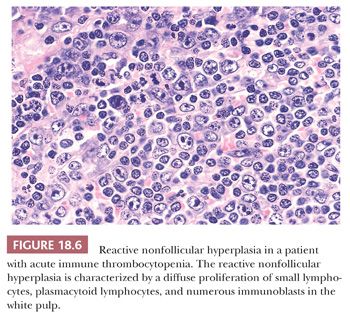
Because reactive nonfollicular hyperplasia is relatively unusual, it may be misdiagnosed as lymphoma. The confusion is not surprising because nests and clusters of immunoblasts often dominate nonfollicular hyperplasia. The observation of an overall heterogeneous lymphocytic population with an orderly range of lymphocytic transformation aids in the distinction from lymphoma. In florid examples of nonfollicular hyperplasia, however, the histologic diagnosis requires supplementation by immunophenotypic markers and frequently by gene rearrangement studies.
MALIGNANT LYMPHOMAS AND RELATED LYMPHOPROLIFERATIVE DISORDERS
The main question posed by clinicians whenever a spleen is removed for “idiopathic splenomegaly” is whether an underlying lymphoma is present (39). In fact, lymphomas restricted to the spleen are uncommon. Most patients who have lymphoma that is first discovered in the spleen have the simultaneous involvement of splenic hilar or abdominal lymph nodes (40). In one series, only 17 of 500 splenectomy specimens (3.4%) with malignant lymphoma were identified as primary lymphoma of the spleen (41). Similarly, only 65 of 1280 (5%) sequential splenectomy specimens were found to contain primary lymphoma (42). Alternatively, occult lymphoma was discovered in 7 of 18 (39%) patients who underwent splenectomy for idiopathic splenomegaly, most of whom had clinical hypersplenism (43), and in a study from Japan that comprised 184 splenic specimens with suspected lymphoma, 115 (62.5%) were determined to be lymphoid neoplasms (44); however, with the exception of large B-cell lymphoma (LBCL), the other newly diagnosed lymphoma cases in that series were discovered in patients with advanced stage disease. LBCL is the most common type of primary splenic lymphoma encountered, with splenic marginal zone lymphoma (SMZL) being second in frequency (42,44). Other series also have documented SMZL as the most frequent form of primary splenic low-grade lymphoma (45,46).
Among patients with known lymphomas, including Hodgkin lymphoma (HL), data from staging laparotomy studies indicate that the spleen is affected in approximately 40% of patients (47); for patients with FL, the prevalence of splenic involvement increases to 56% (48). Although lymphomas develop mainly in the white pulp, the distributional pattern of the various histologic types is diverse yet frequently pathognomonic (49). In the case of many lymphomas, the red pulp is infiltrated secondarily; for some lymphomas, including those composed of large cells, the red pulp is the dominant site of involvement (Table 18.2).

Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
The histologic and immunophenotypic features of chronic lymphocytic leukemia (CLL) are identical to those described for small lymphocytic lymphoma (SLL), so both disorders are categorized as a single entity in the WHO classification (14). Prior to the recognition of SMZL, historically, SLL was the most common type found in patients with primary lymphoma in the spleen (40). Because the majority of patients with this type of lymphoma have stage IV disease, splenic involvement is common (14,50). In cases of CLL/SLL, splenectomy usually is performed because of hypersplenism or autoimmune phenomena and progressive or refractory disease in patients with splenomegaly (51).
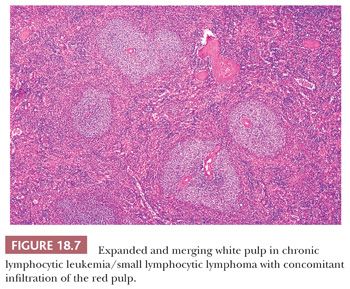
The diagnosis generally is not difficult to determine. In CLL/SLL, the splenic white pulp areas, including the marginal zones and T-cell regions, are randomly expanded and effaced by a proliferation of small, round lymphocytes (49,52). On the cut surface, the white pulp is prominent, and the nodules are asymmetric. Frequently, adjacent expanded white pulp nodules coalesce, and simultaneous diffuse invasion of the red pulp cords and sinuses generally is observed (Fig. 18.7); infiltration of the trabeculae and the subendothelium also occurs. In some cases, the involvement of the red pulp may be so widespread that the white pulp component is obscured. These unusual cases can be difficult to distinguish from hairy cell leukemia (HCL), but immunophenotypic studies, specifically the finding of CD5 and CD23 expression in paraffin sections in CLL/SLL, coupled with a lack of markers associated with HCL—such as tartrate-resistant acid phosphatase (TRAP), DBA.44, cyclin D1, T-bet, CD123, annexin A1, and/or CD103—aid in the diagnosis (53–55).
Other circumstances in which the diagnosis of CLL or SLL of the spleen may be challenging are encountered. One such situation may be the consequence of epithelioid granulomas developing in both splenic white and red pulp. If the granulomas are sufficiently extensive, they may mask the underlying lymphoma (56). Epithelioid granulomas may also manifest in other splenic small B-cell lymphomas, including SMZL, lymphoplasmacytic lymphoma (LPL), and lymphomas that are not otherwise classifiable (52,57). A second source of diagnostic difficulty is encountered in patients whose spleens are of normal weight or minimally enlarged weighing less than 300 to 400 g and removed because of ITP, hypersplenism, or at exploratory laparotomy. Under these conditions, subtle white pulp expansion may be present as a result of a proliferation of small, round lymphocytes (45). Because the small lymphocytes are indistinguishable from normal-appearing reactive lymphocytes, cytologic evaluation is not helpful in this situation. A diagnosis of malignancy may be suspected because of fusion of adjacent white pulp segments with or without secondary spread to the splenic red pulp, and if fresh splenic tissue is available, immunophenotypic analysis, usually by flow cytometry, is required to determine monoclonality and whether CD5 and CD23 are coexpressed (53). If only fixed tissue is accessible, a B-cell immunophenotype with CD5 and CD23 coexpression by immunoperoxidase will facilitate a diagnosis of occult CLL/SLL and separate such cases from reactive lymphoid hyperplasia or another lymphoma. Lymphoid enhancer–binding factor 1 (LEF1) also may prove valuable as a marker of CLL/SLL in contrast to other lymphomas that are composed of small lymphocytes (58).
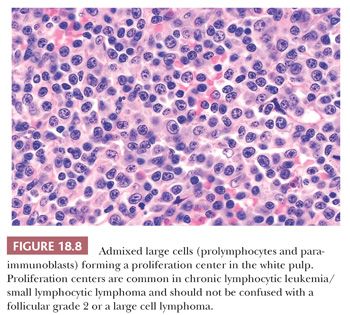
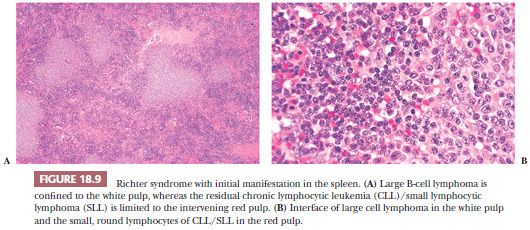
A third problem in establishing a diagnosis of CLL/SLL is seen in cases in which the formation of proliferation centers in the white pulp occurs (52,59). The admixture of larger lymphocytes, specifically prolymphocytes and paraimmunoblasts in the proliferation centers, frequently leads to the misdiagnosis of follicular grade 2 lymphoma or even LBCL (Fig. 18.8); unlike what is found in these other lymphomas, the admixed small lymphocytes in CLL/SLL are round and they are devoid of atypia. The large lymphocytes found in CLL/SLL of the spleen are identical to those reported in their nodal counterparts. If the number of large cells is relatively small, they likely have no bearing on prognosis; however, the identification of focal aggregates of large cells is an uncertain prognostic factor (14,60). On occasion, we have seen spleens in which the number of large cells was so great that it indicated transformation of CLL/SLL to LBCL analogous to Richter syndrome. In splenic Richter syndrome, often a strict compartmentalization of the residual CLL/SLL and the LBCL is present; the small lymphocytes of CLL/SLL are confined to the red pulp, whereas LBCL replaces the white pulp (Fig. 18.9) (45).
Lymphoplasmacytic Lymphoma
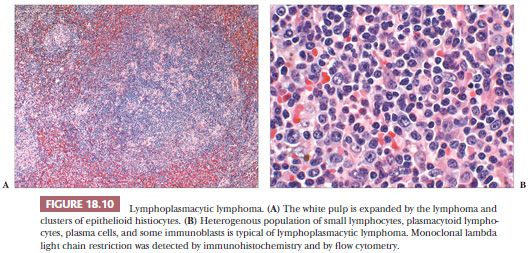
LPL and its clinical counterpart, Waldenström macroglobulinemia, is composed of a variable mixture of small lymphocytes, plasmacytoid lymphocytes, and plasma cells, including cells with Dutcher bodies, generally without CD5 and CD23 expression (14,61). In the spleen, the distributional features are histologically indistinguishable from CLL/SLL. The white pulp is irregularly enlarged as a result of lymphoma. The T-cell areas are often infiltrated, epithelioid histiocytes may be prominent, and nodules may form in the red pulp (Fig. 18.10) (45,49,62). In some cases, the red pulp is diffusely infiltrated with loss of the white pulp and the characteristic nodular pattern of most splenic lymphomas (45,62,63). The absence of proliferation centers and the usual lack of CD5 and CD23 expression serves to distinguish LPL from cases of CLL/SLL with plasmacytoid cells; moreover, the cases of LPL that coexpress CD5 and CD23 usually exhibit partial expression of these antigens in contrast to CLL/SLL (61). LPL is more difficult to distinguish from SMZL, specifically from those cases of SMZL that exhibit plasmacytoid features, because of overlapping immunophenotypic as well as morphologic features (64,65). Both entities are composed of small lymphocytes, lymphoplasmacytoid cells, and plasma cells with similar distributional features in the spleen. Whereas the presence of a pale corona surrounding residual reactive or colonized germinal centers and the identification of marginal zone lymphocytes commonly distinguishes SMZL from LPL, the differential diagnosis can be onerous. Reportedly, one measure of distinction is that the plasma cells in LPL abnormally coexpress CD138 and PAX5 and the median percentage of this coexpression is significantly higher than found in marginal zone lymphoma (66). For certain cases, however, an absolute distinction between LPL and SMZL may not be possible, resulting in a diagnosis of “small B-cell lymphoma with plasmacytic differentiation” (14,61).
Primary plasmacytomas of the spleen are rare and splenic involvement by systemic plasma cell myeloma also is uncommon, although splenomegaly is found in the context of plasma cell leukemia (59,67). Primary splenic plasmacytomas cause massive splenomegaly, and spontaneous rupture may develop (59). Tumor nodules form owing to expanded white pulp segments and nodular aggregates of plasma cells in the red pulp, but, as with LPL, the red pulp may be diffusely infiltrated. The plasma cells range from mature forms to plasmablasts and both binucleated and multinucleated plasma cells are encountered (67). Cytoplasmic monotypic immunoglobulin expression is readily detectable. Distinguishing plasmacytoma dominated by plasmablasts from plasmablastic lymphoma with plasmacytic differentiation is problematic because they share the morphologic and immunophenotypic features of a neoplasm of terminal B-cell differentiation, so that the clinical characteristics remain the only reliable mechanism for accurate classification (68).
Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) typically is a clinically aggressive lymphoma of small lymphocytes that mainly arises from naïve, unmutated, pregerminal center cells localized in primary follicles or the lymphocyte corona or mantle zone surrounding secondary follicles; a subset exhibits somatic hypermutation and arises from antigen-encountered B cells (14,69–71). MCL bears some histologic and immunophenotypic similarities to CLL/SLL, including coexpression of CD5 and CD43, but MCL does not have proliferation centers, and despite some variation, it generally fails to react for CD23 (69–72). Curiously, CD23 expression in MCL is associated with a superior clinical course (72). Of most significance, almost all cases of MCL contain the t(11;14) chromosomal translocation with virtual universal overexpression of cyclin D1 protein (69–71). MCL usually also expresses SOX11 and this neuronal transcription factor identifies rare cyclin D1–negative cases, including the blastoid variant (73). SOX11-negative MCL patients are few, but such patients tend to have leukemic disease without lymphadenopathy but with frequent splenomegaly, lack genomic complexity, and display an indolent clinical course as compared to SOX11-positive patients (74). The inferior survival of the SOX11-positive MCL patients likely is related to the demonstration that SOX11 contributes to tumor development by blocking the terminal B-cell differentiation program in MCL (75).
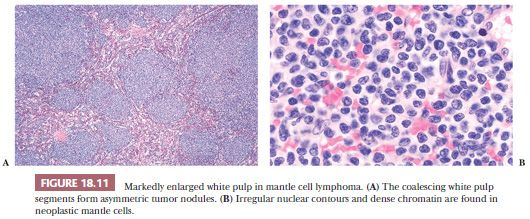
Many patients with MCL have splenomegaly and advanced-stage disease on initial examination (70). In one series, 42% of patients with primary small B-cell lymphoma in the spleen were diagnosed as having the mantle cell type (46). As noted earlier, patients with MCL who present with splenomegaly and without lymphadenopathy generally manifest a leukemic phase, whereas those with the blastoid variant may develop spontaneous splenic rupture (74,76). In some patients with the splenomegalic form of MCL, the peripheral blood and spleen contain nucleolated cells to mimic prolymphocytic leukemia (PLL) (77); unlike PLL, the MCL cases harbor the t(11;14) translocation and express cyclin D1. In the prototypic spleen of patients with MCL, the white pulp is irregularly expanded by a proliferation of small lymphocytes with dense chromatin, indistinct nucleoli, and variable nuclear membrane irregularities (Fig. 18.11) (52). The expanded white pulp may result in confluent tumor nodules or enlarged lymphocytic coronas that surround any residual secondary germinal centers and frequently obliterate the marginal zones. The red pulp cords and sinuses are often infiltrated and the lymphoma frequently forms small nodules in the red pulp (45,46,52). In the splenic cases of MCL that simulate PLL, the lymphomatous cell population is more heterogeneous and includes intermediate-sized cells with coarse, clumped chromatin and single central, small, prominent eosinophilic nucleoli, in addition to the more typical lymphocytes of MCL (77). In some case of splenic MCL, marginal zonelike differentiation—in which cells with more abundant pale-staining cytoplasm are evident—may occur, suggesting SMZL (78); this diagnostic issue is particularly relevant in cases with associated residual germinal centers. The relative monomorphous cytologic appearance, together with cyclin D1 reactivity, should establish the diagnosis of MCL.
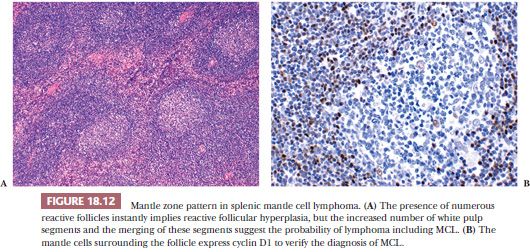
The presence of residual germinal centers in the spleen in cases with the uncommon mantle zone pattern of MCL also may lead to an erroneous diagnosis of reactive follicular hyperplasia. In MCL, however, adjacent white pulp segments coalesce (Fig. 18.12); this observation, when coupled with frequent secondary involvement of the red pulp and nuclear atypia in the proliferating lymphocytes surrounding the germinal centers, serves to distinguish MCL in the spleen from reactive follicular hyperplasia on histologic examination. Immunologic studies demonstrating monoclonality, aberrant CD5 reactivity in B cells, and cyclin D1 expression confirm the diagnosis.
Splenic Marginal Zone Lymphoma
SMZL is a low-grade lymphoma that probably encompasses many primary splenic lymphomas that previously were regarded as CLL/SLL and LPL, rendering SMZL currently the most common primary low-grade lymphoma of the spleen (42,44–46,79). SMZL exhibits histologic heterogeneity, but this lymphoma usually exhibits a marginal zone (monophasic) or a biphasic pattern (80,81). The marginal zone or monophasic pattern is characterized by expansion of the marginal zone of the splenic white pulp due to a proliferation a marginal zone lymphocytes, together with varying numbers of larger blast-like cells and plasma cells (46,79–84). Marginal zone lymphocytes are small to medium-sized cells with ovoid to reniform-shaped nuclei and a characteristically abundant pale-staining cytoplasm that differentiates them from adjacent mantle zone lymphocytes. Splenic marginal zone lymphocytes resemble monocytoid B cells, and cases of disseminated nodal MZL and extranodal MZL of mucosa-associated lymphoid tissue (MALT) have in fact been reported in the spleen, where they involve the splenic marginal zone and are indistinguishable from primary SMZL (85,86).
In SMZL, the marginal zones are expanded and the neoplastic marginal zone lymphocytes surround, encroach upon, and frequently invade attenuated rims of mantle zone lymphocytes and secondary germinal centers, if these are present (Fig. 18.13). This marginal zone (monophasic) pattern may be observed in both massively enlarged spleens and spleens of normal weight in the presumed early phase of the lymphoma (87). In the latter case, replacement of the T cells in the periarteriolar lymphoid sheaths by the neoplastic B cells of SMZL often occurs. In contrast to SMZL in low-weight spleens, in markedly enlarged spleens, residual mantle zone lymphocytes and germinal centers usually are completely obliterated and are replaced by neoplastic marginal zone lymphocytes forming relatively monomorphous, coalescent tumor nodules in the white pulp. Red pulp infiltration usually is present, and, in a few cases, red pulp involvement is extensive leading to almost total destruction of the white pulp with only small, residual, micronodular white pulp segments remaining (80). A predominant diffuse red pulp pattern of infiltration has been described in cases thought to represent SMZL, but the current WHO classification places such cases in the provisional category of splenic diffuse red pulp small B-cell lymphoma (SDRPL) (14,88).
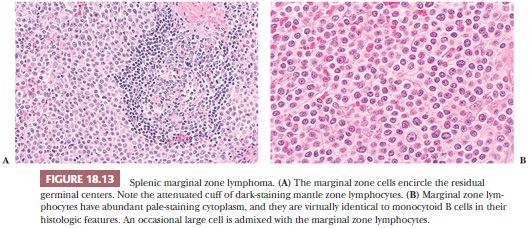
In other cases, SMZL has a distinct lymphoplasmacytic component; such cases often are associated with a monoclonal gammopathy and autoimmune hemolytic anemia (64,89); as discussed earlier, the lymphoplasmacytic form of SMZL is exceedingly difficult to differentiate from splenic LPL (64,65). In still other cases of SMZL, increased numbers of blasts or large cells are found, and frank transformation to LBCL may develop (82,89–91).
The biphasic pattern of SMZL comprises cases in which the white pulp manifests a prominent central core of small lymphocytes, similar to the cells of MCL, that is surrounded by a peripheral zone of marginal zone–type cells (Fig. 18.14); both zones are considered part of the same neoplastic clone (81,89,92,93). The inclusion of biphasic cases in an expanded definition of SMZL arose out of a review of the splenic histologic findings of 37 cases documented as splenic lymphoma with villous lymphocytes (SLVL) (92). In that study, all of the cases of SLVL, including those with a biphasic appearance, were interpreted as being identical to SMZL; these cases currently are categorized as a leukemic form of SMZL (14,89,92,93).
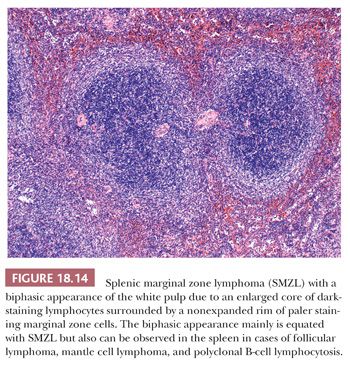
Leukemic SMZL is a chronic lymphoproliferative disorder that predominantly develops in older adult men. Consistent massive splenomegaly and circulating atypical lymphocytes with cytoplasmic villous projections that often are localized to one pole of the cell are observed (89). A monoclonal spike is found in the serum or urine in about one-third of patients. About 15% of patients initially thought to represent SLVL have translocation of t(11;14)(q13;q32) and rearrangement of the bcl-1 locus with increased expression of cyclin D1; the latter patients exhibit a more aggressive clinical course and actually are leukemic MCL cases simulating leukemic SMZL, including the biphasic pattern (46,78,94,95). The biphasic pattern also may occur in splenic FL and in cases of marginal zone hyperplasia, such as found in ruptured spleens (3). One morphologic sign that an expanded marginal zone is neoplastic is the frequent coalescence of adjacent marginal zone segments in SMZL as opposed to the absence of merging of bordering zones in marginal zone hyperplasia; nonetheless, this distinction on histologic examination can be formidable, and immunoglobulin gene rearrangement studies may be required for an absolute diagnosis (96). One form of marginal zone hyperplasia develops in enlarged spleens derived from patients with persistent polyclonal B-cell lymphocytosis, an uncommon benign condition typified by a stable polyclonal CD19-positive, CD5-negative lymphocytosis (97,98). In persistent polyclonal B-cell lymphocytosis with splenomegaly, the splenic marginal zones are expanded with a biphasic appearance and with associated striking infiltration of the red pulp cords and sinuses by small lymphocytes. The histologic distinction from SMZL is demanding, but unlike SMZL, in persistent polyclonal B-cell lymphocytosis, the infiltrating B lymphocytes are polyclonal (97,98).
SMZL is a B-cell neoplasm expressing various B-cell antigens, IgM and IgD monoclonal immunoglobulin, and bcl-2 protein; CD5, CD10, CD23, CD43, and cyclin D1 generally are not reactive (46,79,69,81,84,89–93). The usual absence of CD5 and CD43 expression, as well as cyclin D1, eliminates CLL/SLL and MCL from the diagnosis, and lack of CD10 excludes FL (46,64,78,95). Cases of SMZL that express CD5 exhibit higher lymphocytosis and more frequent diffuse bone marrow infiltration but otherwise are no different than the more prevalent CD5-negative cases (99).
Despite similar cytologic features, immunophenotypic profiles, and a shared designation as “marginal zone,” SMZL differs genetically from its extranodal namesake. Similar to node-based MZL, but unlike extranodal MZL of MALT, the splenic cases do not have detectable t(11;18) translocations or API2–MALT1 fusion transcripts (100). Patients with SMZL, however, display a variety of complex karyotype abnormalities with frequent gains of 3/3q and 12q as well as genetic losses, specifically 7q and 6q deletions (101). Deletion of 7q is a characteristic feature of SMZL and its discovery can aid in the differential diagnosis as it is only also detected in some cases of splenic B-cell lymphoma/leukemia, unclassifiable (81,102). Recently, mutations in NOTCH2, a gene required for marginal zone B-cell development, has been discovered in 20% to 25% of cases of SMZL (103,104). NOTCH2 mutations appear exclusive to SMZL and are not present in other B-cell lymphomas and leukemias. Moreover, NOTCH2 mutations are an adverse clinical finding and associated with increased risk of relapse, histologic transformation, and death (103). Complexity of the karyotype, 14q aberrations, and TP53 deletions also are indicative of a poor prognosis (101). In general, however, SMZL is an indolent lymphoma, and in a report of 309 patients, the 5-year cause-specific survival rate was 76% (105). Hepatitis C virus may play a role in the lymphomagenesis of SMZL, and some patients benefit from antiviral therapy (106).
Splenic Diffuse Red Pulp Small B-Cell Lymphoma
Although clearly not a disorder of the white pulp, SDRPL is a provisional category of indolent lymphoma in the 2008 WHO lymphoma classification scheme (14). This form of lymphoma encompasses the cases that were formerly designated as the diffuse red pulp variant of SMZL and more recently reported as splenic red pulp lymphoma with numerous basophilic villous lymphocytes (80,84,88,107). Such lymphomas are associated with peripheral blood involvement, often with lymphocytes containing villous projections that are unevenly distributed and with a polar position, as well as bone marrow involvement (107). In the spleen, the red pulp cords and sinuses are diffusely infiltrated by small to medium-sized, generally round lymphocytes with clumped chromatin, inconspicuous nucleoli, and pale cytoplasm (Fig. 18.15). Similar to HCL and hairy cell leukemia variant (HCL-V), pseudosinuses and red cell lakes may be present and the white pulp usually is atrophic or obliterated (93). The neoplastic lymphocytes express CD20 and DBA.44, but unlike HCL, they are CD25-negative and usually CD103, CD123, and annexin A1–negative (14,107,108). Parenthetically, a case of SDRPL with unexplained annexin A1 reactivity has been reported to emphasize that no single molecule is absolutely exclusive to a disease (109).
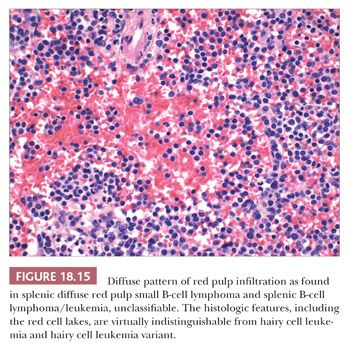
Similar to SMZL, SDRPL does not express CD5, CD10, or CD23 and by flow cytometry, CD11c usually is moderately expressed (110). The cells are IgG-positive and may also express IgD. TRAP staining may occur, and cases often show IgH mutations (107–109). Clonal cytogenetic alterations generally are absent, but this lymphoma may exhibit chromosome 7q deletion, complete trisomy 18, and partial trisomy 3q, but not t(11;14) (107). As opposed to HCL, BRAF (V600E) mutation has not been described in SDRPL (109–111). Although SDRPL is a provisional category in the WHO system, and although usually CD103-negative, it displays considerable overlap with HCL-V so that additional cases and further investigations are required to further delineate these uncommon lymphomas and/or leukemias (112).
Follicular Lymphoma
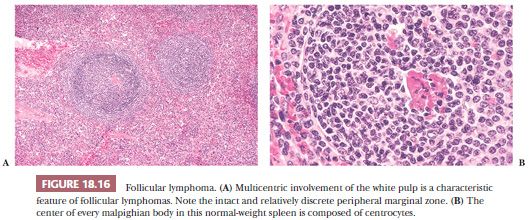
Involvement of the spleen by lymphomas with a follicular architecture in lymph nodes is morphologically distinctive. Because these lymphomas usually involve multiple lymphoid sites initially, FL in the spleen, specifically the grade 1 and grade 2 types, similarly embrace virtually every splenic lymphoid site in a multicentric pattern (1,113). With extensive disease, the splenic cut surface is peppered by uniformly expanded white pulp nodules that allow ready diagnosis on microscopic examination. The uniform multicentric pattern, when it is coupled with grade 1 or grade 2 cytology, generally implies, but does not absolutely equate with, a follicular architecture in nodes. In the spleen, the neoplastic follicles frequently contain extracellular hyaline deposits (52).
The era of splenectomy for pathologic staging of non-HL long has passed, but examination of those spleens demonstrated that almost one-third were involved by lymphoma, although the spleens were not palpable on clinical evaluation (48). The spleens usually were of normal weight and, even though obviously expanded white pulp was absent on either gross or low-power microscopic examination, a detailed pathologic analysis demonstrated that practically every white pulp segment contained a central core of neoplastic cells, usually centrocytes (Fig. 18.16) (113). In this context, grade 2 cases are more difficult to diagnose with confidence because they resemble reactive follicular hyperplasia (1). Such lymphoma cases, however, usually lack tingible body macrophages; the mantle zone is often diminished or obliterated; no associated plasmacytosis in the red pulp exists; and, paradoxically, the distribution in the white pulp is usually more homogeneous than that observed in hyperplasia. Immunologic markers, such as bcl-2 protein reactivity, are an important adjunct for reaching the diagnosis. The pattern of subtle involvement of the splenic white pulp by FL has come to the fore more recently and referred to as the architectural preservation pattern (114). Involvement of the spleen by FL with architectural preservation may be focal and such cases parallel the cases of FL in situ reported in lymph nodes (114,115). One extraordinary case of composite FL and MCL in the spleen with each exhibiting an in situ component has been described (116).
For cases of FL that present primarily with splenic enlargement, the multifocal pattern results in architectural abnormalities with frequent micronodules that efface the splenic red pulp, although the marginal zones at the periphery of the nodules generally are preserved (117,118). In this setting, two variants of splenic FL were reported—one was similar to classic FL with grade 1 to 2 morphology, CD10 and BCL2 expression, t(14;18) positivity, and low proliferative activity, whereas the second tended to be more morphologically high-grade with variable expression of CD10 and BCL2, t(14;18) negativity, and a higher proliferative rate (117). The second group, moreover, were more frequently primary lymphomas of the spleen and were at greater risk for transforming to LBCL.
In many spleens involved by FL, regardless of weight, the marginal zones often are conspicuous and may resemble SMZL (1,45,52,78,117,118). The splenic cases containing FL with prominent marginal zone differentiation are similar to cases in lymph nodes (119,120). Those cases with concurrent FL and marginal zone differentiation usually exhibit coalescence of adjacent marginal zones. In addition, the central follicular component of the expanded white pulp strongly expresses bcl-2 protein in comparison to the weak reactivity of the peripheral marginal zone component; the t(14;18) translocation may be detected (119,120). Molecular genetic studies, however, demonstrate that both components have the same clonal sequences and, together with the BCL2 rearrangements, indicate that such cases are of follicular center cell origin (121).
Large B-Cell Lymphoma
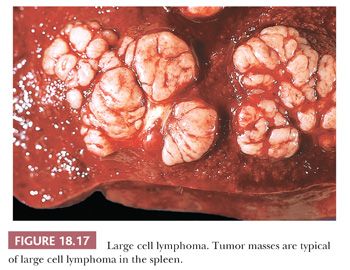
LBCL is the most common primary lymphoma in the spleen (42,44). Similar to SMZL, hepatitis C virus may have a role in the lymphomagenesis of splenic LBCL (106,122). Classically, it appears as a large tumor mass or a series of large, confluent nodules extensively replacing the splenic parenchyma and secondarily invading the red pulp (Fig. 18.17) (1,41,123,124). The lymphoma frequently breaches the capsule to invade contiguous organs and may present with spontaneous splenic rupture (125,126). The distribution appears completely random, and the neoplastic cells are often adjacent to uninvolved segments of white pulp (45,49).
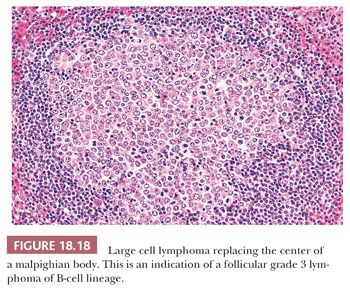
Primary splenic LBCL cases include not only those with large coalescent tumor masses but also those with multiple, discrete monomorphous nodules in the centers of the white pulp (Fig. 18.18); an association with CLL/SLL, FL of grade 1 or 2 type, or SMZL; and a centroblastic or immunoblastic plasmacytoid cytology (123,124). Some examples of LBCL may manifest initially in the marginal zone, resulting in the formation of crescent-shaped infiltrates around intact follicles (59,127).
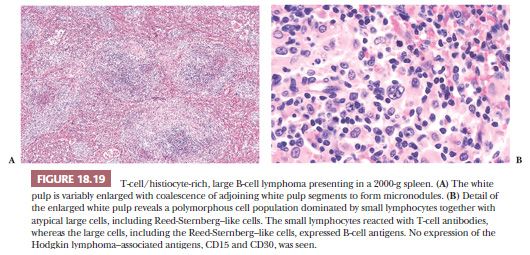
Although these histologic features provide guidelines, immunologic analysis is mandatory for an absolute discrimination between B-cell and T-cell large cell lymphomas in the spleen. For example, T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) may involve the spleen (5,124,128,129). THRLBCL cases in which the spleen is the initial site of disease mimic reactive inflammatory lesions as well as peripheral T-cell lymphoma (PTCL) and HL (Fig. 18.19) (5,128,129). THRLBCL forms a series of micronodules engaging the white pulp. The nodules are composed of scattered large B cells set among abundant T cells and histiocytes. Paraffin section immunophenotypic studies are invaluable in these cases and they demonstrate that the micronodules comprise numerous CD3-reactive T cells, CD68-positive histiocytes, and randomly dispersed large cells expressing CD20 with immunoglobulin light chain restriction (128). The prognosis in these cases is poor.
The spleen also is a repository for rare cases of LBCL that primarily affect the splenic red pulp with diffuse infiltration of the cords and, occasionally, the sinuses (123,124,130,131). In addition to B-cell antigens, the diffuse red pulp splenic LBCL cases may coexpress CD5 (130,131). If the large B cells are predominately distributed in the red pulp sinuses, such cases may represent a version of intravascular LBCL and, principally in Asia, may be affiliated with a hemophagocytic syndrome and aggressive clinical behavior (132,133). The diagnosis of splenic involvement by intravascular LBCL requires confirmation by the detection of extrasplenic intravascular lymphoma (134). The Asian form of intravascular LBCL shares features, including hemophagocytosis and an aggressive clinical course, with a type of LBCL that initially presents in bone marrow, liver, and spleen but without an intravascular component (135); the splenic lymphoma in this form of LBCL consists of a series of micronodules in the red pulp that are composed of centroblasts with admixed inflammatory cells. The division of primary LBCL of the spleen into germinal center (CD10+, bcl-6+, MUM1−) and nongerminal center (CD10−, bcl-6+/−, MUM1+) types and the prognostic relevance of MYC/bcl-2 protein coexpression in such cases has not yet been analyzed (136,137).
T-Cell Lymphoma
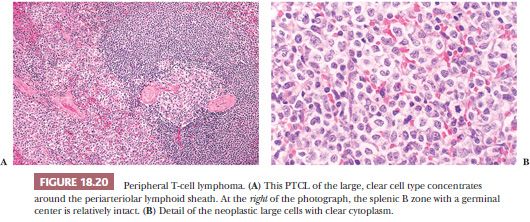
Splenic lymphomas of peripheral T-cell origin, including those with a mixed cellular composition and those with epithelioid histiocytes (Lennert lymphoma), are uncommon in contrast to those of B-cell lineage, and they may be confined to the periarteriolar lymphoid sheaths (Fig. 18.20) and/or marginal zones (49,59,123,138,139). At the latter site, epithelioid histiocytes form a ringlike array around the white pulp. Cases without epithelioid histiocytes may form seemingly indiscriminate tumor nodules. Among immunologically verified PTCLs in the spleen, histologic features favoring a T-cell phenotype include not only epithelioid histiocytes and confinement of the lymphoma to the splenic T zones (periarteriolar lymphoid sheaths) or periphery of the marginal zones but also clear cell or polymorphous cytology (123). More commonly, however, neoplastic T cells diffusely invade the red pulp without the involvement of white pulp or obvious formation of tumor masses (Fig. 18.21) (139,140); many such cases are of the hepatosplenic T-cell type (141).
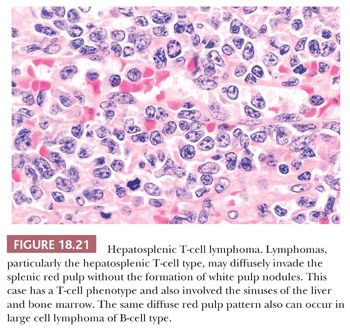
Hepatosplenic T-cell lymphoma (HSTL) is a rare, clinically aggressive cytotoxic T-cell lymphoma sometimes arising in immunocompromised patients, in which neoplastic medium-sized to large lymphocytes infiltrate the splenic sinuses and, occasionally, the cords as well as the sinuses of the liver and bone marrow (139–146). The HSTL cases may show extensive tumor necrosis and hemophagocytosis. Phagocytosis may be performed by the neoplastic-appearing cells or by true, albeit reactive, histiocytes (139,140,143–146). The immunophenotype is characteristic in that, in addition to T-lineage markers, the neoplastic lymphocytes commonly express natural killer (NK) cell antigen CD56, and the cytotoxic granule–associated protein TIA1, but do not express CD4, CD8, and only occasionally EBV sequences (139,142–146). Most cases are of γδ T-cell type and display an isochrome 7q (142–146); however, identical-appearing HSTLs of αβ type develop and those cases may also have isochrome 7q (146,147). Compared to other T-cell lymphomas, HSTL exhibits a distinct molecular signature regardless of the T-cell receptor lineage (148).
Other T-cell lymphomas found in the spleen are angioimmunoblastic T-cell lymphoma (AILT), anaplastic large cell lymphoma (ALCL), S100 protein–positive PTCL, and mycosis fungoides (MF). Although splenomegaly is reported in about half the patients with AILT, descriptions of splenic AILT are uncommon, and, as in lymph nodes, AILT may be easily confused with nonfollicular atypical hyperplasia (149,150). In AILT, white pulp expansion may be accompanied by increased vascularity and a concomitant polymorphous infiltrate into the red pulp (150). Unless cases of presumptive AILT are associated with cytologic atypia or cellular monomorphism, distinguishing them from an atypical form of reactive lymphoid hyperplasia is problematic and gene rearrangement studies may be required (151). The neoplastic T cells in AILT arise from follicular helper T cells and express markers such as CD10, bcl-6, CXCL13, PD-1, and/or ICOS that may be used to distinguish AILT from most other PTCLs (14,152,153). Primary splenic presentation of ALCL is rare, but if the spleen is involved, ALCL usually forms fleshy tumor nodules (139). ALCL cases have been described in association with spontaneous splenic rupture (154) and with symptoms of a splenic abscess (155). S100 protein–positive PTCL is an uncommon form of PTCL (139); in the spleen, this lymphoma exhibits diffuse red pulp infiltration by medium-sized or large lymphocytes. In addition to expressing T-lineage markers and S100 protein, the neoplastic lymphocytes are frequently CD56-positive (139).
MF is another form of T-cell lymphoma that occasionally manifests in the spleen. In a pilot study during the period of staging laparotomy, most cases of MF were discovered to concentrate in the splenic T-dependent zones, but, like aggressive PTCL, heterogeneity is seen among these cases (156). For example, some cases of MF form irregular nodules throughout the white and red pulp, whereas other cases diffusely infiltrate the red pulp cords and sinuses. As opposed to the more standard cases of HSTL, splenic PTCL, and splenic MF that exhibit nuclear atypia and an aggressive clinical course, occasional examples of PTCL that diffusely involve the splenic red pulp are composed of small lymphocytes with limited mitotic activity (157); these cases have a low Ki-67 index, commonly display a phenotype that corresponds to central memory T cells (CD45RO+, CCR7+), and are clinically indolent.
Hodgkin Lymphoma
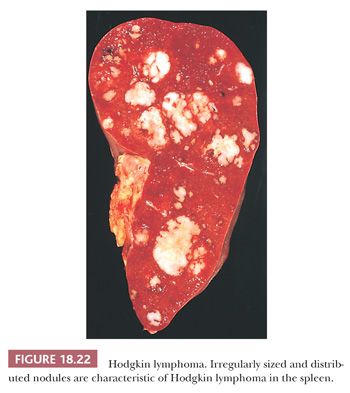
HL with an initial diagnosis in the spleen is rare, and staging laparotomy with splenectomy is no longer a routine procedure for this lymphoma (158). Characteristically, spleens involved by HL contain scattered haphazard nodules with a miliary distribution (Fig. 18.22), but, occasionally, only a solitary focus as small as 1 mm in diameter may be present (159). These foci may not be noticed unless the spleen is meticulously sectioned.
The diagnosis of HL in the spleen is dependent on the identical histologic criteria that are employed in assessing the lymph nodes; however, even in advanced cases, Reed-Sternberg cells may be difficult to identify in the spleen. Similar to the lymph nodes, focal or early lesions are localized in the periarteriolar lymphoid sheaths and marginal zones (160). That epithelioid sarcoid-type granulomas may be seen in up to 9% of HL cases and that they may even form macroscopic nodules has been documented (161). The discovery of granulomas does not indicate involvement of the spleen by HL, because these granulomas may be found in cases with no association with HL.
Subclassification of HL in the spleen is optional. Moreover, because some cases of mixed cellularity HL have considerable fibrosis, whereas cases of known nodular sclerosis HL may lack apparent lacunar cells in the spleen, subclassification may be inaccurate (1). Because subclassification of HL is not a factor in therapy, I tend not to subclassify (47).
Prolymphocytic Leukemia
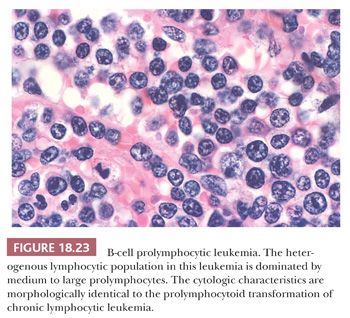
Splenomegaly is the major physical finding in PLL, a rare malignancy associated with prominent splenomegaly that by definition must contain 55% or more prolymphocytes in the peripheral blood (14). The general histologic distribution of PLL in the spleen is similar to that of CLL/SLL, but many more intermediate-sized to large prolymphocytes in which the heterochromatin is more dispersed are observed and small nucleoli are present (Fig. 18.23) (162,163). Paraimmunoblast-type cells also are evident. Making the histologic distinction between PLL and prolymphocytoid transformation of CLL/SLL is tenuous without a clinical history (59). Most cases of PLL have a B-cell phenotype, and, unlike CLL/SLL, strong expression of surface immunoglobulin and usually no reactivity for CD5 are found (164). B-cell prolymphocytic leukemia (B-PLL) is biologically heterogeneous with respect to IgVH mutations, ZAP-70, and CD38 expression, exhibiting a pattern that is reported to be distinct from that of other lymphoproliferative disorders (165). As discussed earlier, however, prolymphocyte-like cells may be observed in the splenomegalic variant of MCL and, in contrast to B-PLL, the MCL cases express cyclin D1 as well as seat the t(11;14) translocation (77). In addition, prolymphocytoid transformation with greater than 55% prolymphocytes in the peripheral blood may develop in cases of splenic B-cell lymphoma, specifically SMZL and SDRPL (166). Such cases raise the question as to whether B-PLL is actually a discrete clinicopathologic entity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


