
SPINDLE CELL TUMORS
18.5 Desmoplastic Fibroma of Bone
18.6 Undifferentiated High-Grade Sarcoma of Bone
Fibrous dysplasia (FD) is a benign dysplastic process, which may involve one (monostotic) or more (polyostotic) bones. This lesion tends to occur in children and younger individuals with a peak incidence between 5 and 20 years of age. Both sexes are equally affected. Individuals with the polyostotic form of disease tend to present at a younger age, usually before 10 years. The monostotic form of FD is more common and accounts for about 75% of all cases. The polyostotic form of the disease has two distinctive presentations: a monomelic variant confined to one extremity and ipsilateral pelvis or the polymelic variant, which tends to involve several different bones. Polyostotic FD is associated with both McCune-Albright and Mazabraud syndromes.
FD tends to involve the long bones, ribs, as well as the craniofacial bones. Monostotic lesions are frequently asymptomatic and are often incidental findings on imaging studies performed for other reasons. Symptomatic lesions can present with localized pain or, occasionally, pathologic fracture. FD of the femoral neck is particularly prone to fracture due to structural weakness in a high load-bearing region of the bone.
The classical feature of FD on imaging is the presence of a lucent lesion with a “ground glass” type of appearance (Figure 18.1.1). Often there is a distinct rim of reactive bone surrounding an area of hazy lucency. There may be extensive distortion of the normal bone architecture due to stress on the weakened bone. In the proximal femur, this often leads to a “Shepherd’s crook” type of malformation. In general, FD does not show a periosteal reaction unless there has been a pathologic fracture.
Treatment of FD is usually conservative. Asymptomatic lesions that are not at risk for fracture can simply be followed by imaging. Lesions that tend to compromise the structural integrity of the bone can be curettaged and grafted. Although FD is considered benign, there are rare reports of malignant transformation, usually involving the development of osteosarcoma, fibrosarcoma, or chondrosarcoma. Although the exact incidence of this event is not known, it is estimated to occur in less than 4% of the cases.
FD has been linked to activating mutations in the Gs alpha subunit gene (GNAS1) located at 20q13.2. This mutation occurs in a mosaic distribution pattern early on in embryogenesis. The extent and severity of FD in any one individual relates to the extent and distribution of the mutated cells.
HISTOPATHOLOGY
FD is usually a well-circumscribed and delineated lesion. It is composed of a background fibrous component as well as an osseous component. The latter is characterized by irregular thin trabeculae of woven bone arranged in a variety of patterns (Figure 18.1.2). The trabeculae have often been described as “alphabet soup” or “Chinese characters” in configuration. They are occasionally anastomosing, and consist of rounded or curvilinear fragments of bone with “C” and “S” shapes (Figure 18.1.3). Variations on the appearance of the bone component include the presence of psammomalike or cementoid bodies (Figure 18.1.4). The latter are particularly more prevalent in FD of the facial bones.
FD is a dysplastic process. As such, osteoid forms directly from the fibroblastic background. In FD, there should not be prominent osteoblastic rimming associated with the bone fragments (Figure 18.1.5). Some older FD lesions can show irregular calcification of the osteoid.
The background component of FD can be variably cellular and will occasionally show evidence of degenerative change. The latter includes the presence of foamy macrophages, myxoid change, and microscopic as well as macroscopic cystic degeneration (Figure 18.1.6). Extensive cystic change can sometimes lead to a false impression of aneurysmal bone cyst.
Occasionally FD shows a prominent cartilaginous component. Fibrocartilaginous dysplasia tends to preferentially involve the femur, and the presence of extensive cartilage can sometimes be identified on imaging studies. In this variant of FD, cartilaginous nodules are often distributed throughout the lesion. They are frequently encircled by a rim of bone, either woven or sometimes lamellar in character. The chondrocytic component tends to be bland, without hypercellularity or cytologic atypia.
CYTOLOGIC FINDINGS
FD tends to yield very poorly cellular aspirates. Aspirates tend to consist entirely of small fragments of bland stroma, which are not specific for diagnosis. Cystic FD may yield straw-colored fluid with macrophages.
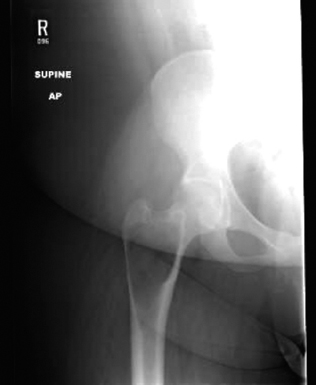
FIGURE 18.1.1 On plain films, FD is a lucent lesion with an interior described as “ground glass.” This is an example of FD in the metaphysis of the proximal femur. The lesion is expansile, but the cortex is intact.
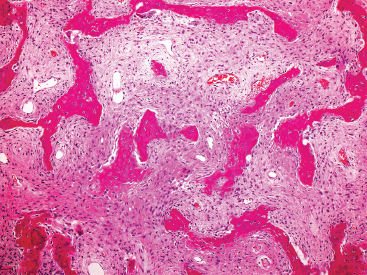
FIGURE 18.1.2 Histologically, FD is composed of two elements: a bland spindle cell proliferation in the background and superimposed irregular fragments of osteoid.
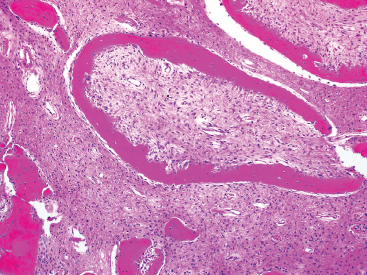
FIGURE 18.1.3 The osteoid of FD is often arranged in C- or S-shaped configurations. This pattern is often described as “alphabet soup.” Note also the retraction artifact adjacent to the osteoid fragment at the top of the picture.
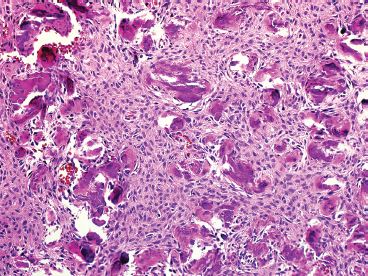
FIGURE 18.1.4 Psammoma-like or cementoid configurations of osteoid are often seen in FD that occurs in the craniofacial bones.
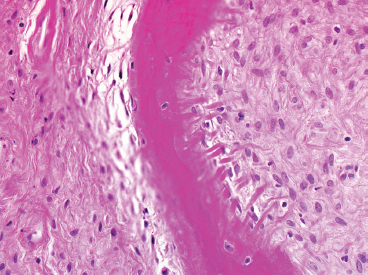
FIGURE 18.1.5 A high-power view of one of the osteoid fragments in FD. Note the absence of osteoblasts lining the lesional osteoid. Instead, FD is a metaplastic process, and osteoid is produced directly from the fibroblastic tissue.
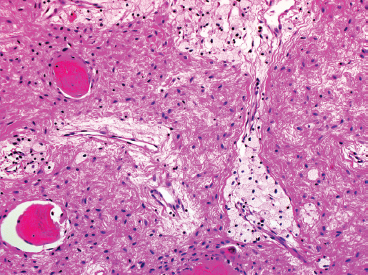
FIGURE 18.1.6 Cystic degeneration is common in FD, particularly in older lesions. One of the earliest histologic manifestations of this process is the presence of focal foamy macrophages within the lesion.
Nonossifying fibroma (NOF) is a common, benign neoplasm of bone. NOF is also frequently called “metaphyseal fibrous defect,” “fibroma,” “benign fibrous histiocytoma of bone,” and “fibrous cortical defect.” These are all synonyms for a benign, probably reactive lesion. NOF is often an incidental finding, and its true incidence is unknown. Clinically, NOF tends to present in children who are still undergoing skeletal growth with a peak incidence between 10 and 20 years of age. The most common locations for NOF include the metaphyseal regions of the distal femur, proximal fibula, and proximal and distal aspects of the tibia. Older patients with a “healed” or regressing NOF often have diaphyseal lesions. Patients are usually asymptomatic, but can occasionally present with localized pain. More commonly, pathologic fracture occurs through the structurally weak bone affected by NOF. Most cases of NOF are solitary, but multiple lesions occur in Jaffe-Campanacci syndrome. The latter includes multiple osseous lesions, café au lait-like skin spots, cryptorchidism, and developmental delay.
On imaging, NOFs are fairly distinctive, and a definitive diagnosis is often made by radiographic features alone. NOFs are eccentrically placed and usually positioned with the long axis of the lesion in line with the long axis of the host bone (Figure 18.2.1). They tend to be cortical based, but very large lesions can obscure the relationship with the underlying normal bone. NOFs are often very lytic in their “active” phase and always have very sharp borders, often with a slight rim of sclerosis.
NOFs will eventually regress over time. Small lesions that do not compromise the structural integrity of the bone can simply be followed with serial imaging and observation. Larger lesions that pose a threat of fracture can be curettaged and grafted.
HISTOPATHOLOGY
NOFs tend to be very cellular and are composed of numerous plump spindled cells. They are classically arranged in a storiform pattern (Figure 18.2.2). Individual cells tend to be relatively monotonous with no pleomorphism. NOFs frequently contain osteoclast-type multinucleate giant cells (Figures 18.2.3 and 18.2.4). These are usually scattered throughout the lesion. Other prominent features include foci of macrophages with a foamy appearance, pigment-laden macrophages, and small foci of inflammation (Figures 18.2.5 and 18.2.6). Mitotic figures are often numerous in NOF. In older areas, the background stroma may become more fibrotic and sclerotic. In addition, “maturing” NOFs tend to contain foci reactive bone, often accompanied by osteoblastic rimming.
CYTOLOGIC FINDINGS
Aspirates of NOF tend to be moderately cellular and composed of a mixture of spindled cells, macrophages, and occasional osteoclast-type giant cells. The spindle cell population is usually predominant and is identifiable as small clusters of cells as well as individual cells. Individual cells have open chromatin and smooth nuclear contours.
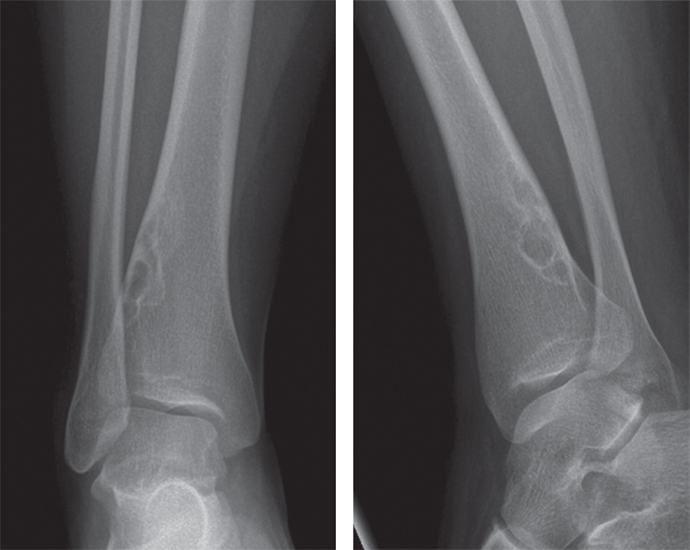
FIGURE 18.2.1 Imaging of NOF shows an eccentrically placed, lobulated lesion of the metaphysis with sclerotic borders.
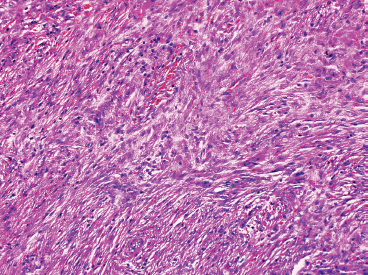
FIGURE 18.2.2 NOF is composed of bland spindled cells arranged in a storiform pattern. There is also admixed mild chronic inflammation.
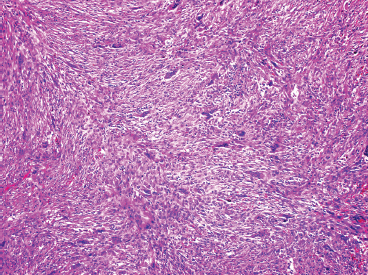
FIGURE 18.2.3 Osteoclast-type multinucleate giant cells are often prominent within NOF.
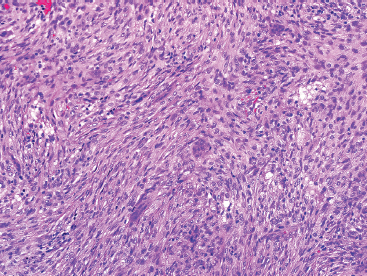
FIGURE 18.2.4 Higher power of NOF showing a bland spindle cell population as well as rare giant cells.
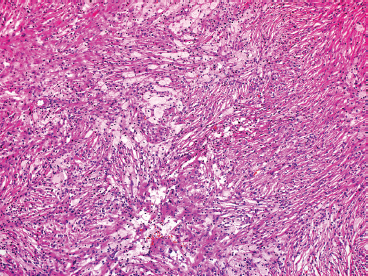
FIGURE 18.2.5 Numerous foamy histiocytes are present.
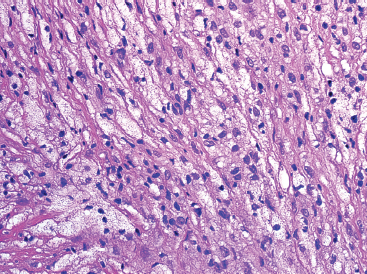
FIGURE 18.2.6 In older lesions, foamy macrophages often predominate over the spindle cell component.
Osteofibrous dysplasia (OFD) is a rare, benign, slow-growing lesion of bone that accounts for less than 1% of all bone lesions. It is sometimes referred to as Camapanacci disease, after one of the authors who first described and characterized this lesion. OFD is more common in boys and tends to peak in mid-childhood (median age 9.5 years) to early adolescence. Rare cases have been reported in younger children including neonates. The proximal or mid-tibia is by far the most common site of involvement, with 85% of all cases occurring at this site. Tibial lesions may show involvement of the adjacent fibula. The second-most common sites include the radius and ulna. Common symptoms include painless swelling and often deformity of the affected region.
On imaging, OFD appears as a longitudinally oriented lesion with multiple lucencies. The linear extent of OFD can be impressive, often involving a very long segment of bone. OFD tends to be centered on the cortical region of the bone, but there is often extension into the adjacent medullary cavity. The affected cortex is often thinned, and a bowing abnormality may develop (Figure 18.3.1). The medullary aspect of the lesion is often characterized by dense sclerosis.
The prognosis of OFD is excellent. After an initial growth period, OFD tends to stabilize, usually around 15 years of age. This is often followed by spontaneous regression of the lesion over time. Given the natural history of OFD, appropriate management is usually a “watch and wait” type of strategy. However, when extensive deformity or pathologic fracture is present, the lesion should be excised. Because of the substantial clinical and radiographic overlap with adamantinoma, OFD is occasionally biopsied for confirmation of diagnosis. To date, the few cases of OFD that have been subjected to cytogenetic analysis have shown trisomies, most commonly of chromosomes 7, 8, 12, and 21.
HISTOPATHOLOGY
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


