Chapter 6 Providing drug therapy to pregnant or lactating women poses a unique challenge for health care practitioners. Because these patients represent a state of duality, the clinician who prescribes a drug for them must always consider the impact of that treatment on the developing fetus or infant. Hence the benefit of any drug to a pregnant patient must be carefully weighed against the potential teratogenic risk to the fetus. In the lactating mother, the choices between premature weaning (before the age of 1 year), temporarily interrupting breastfeeding, or breastfeeding while taking medication must be carefully weighed against the benefits of breastfeeding and the deleterious effects of early weaning. Because controlled clinical trials cannot be conducted in pregnant women, literature support of current practice is based on historical data, case reports, and animal research trials. Much of the literature in this area is based on case reports and drug research, but it may not be evidence based in nature (AAFP, 2002; AAP, 2001; McPhee, Papadakis, & Rabow, 2012). During pregnancy, the clinician must be aware of the changing physiologic characteristics of the patient throughout gestation, as well as those of the growing fetus. Additionally, although multiple factors may affect the teratogenicity of a drug, one of the most important is the timing of the drug exposure. Three stages of development are generally considered when the teratogenic potential of a drug is assessed (Briggs, 2011; Koren, 2006): the 2 weeks after conception and before implantation (the preimplantation phase), the embryonic period (weeks 3 through 8), and the fetal period (weeks 9 through delivery). In the past, clinicians have overestimated the ability of the placenta to protect the fetus, and in fact, the term placental barrier is a misnomer in that the placenta allows the crossing of most drugs and dietary substances. For the breastfed infant, the provider must understand the dynamics between the mechanisms of the drug’s entry into mother’s milk and what happens once the infant ingests it, taking into account the infant’s age, health status, and ability to metabolize and excrete the medication. Primarily, medication transfers into human milk through a concentration gradient that allows passive diffusion of free (non–protein-bound) and nonionized medication. Different choices and greater caution may be needed in infants younger than 1 week old, premature infants, and compromised infants with health problems because they may have a lessened ability to tolerate, metabolize, or excrete medications. Finally, more medication transfer occurs during the early postpartum period because of large gaps between the mammary alveolar cells, but less transfer occurs during the weaning process because of a decrease in the milk supply caused by nursing sessions that are shorter or fewer (Koren, 2006). Overall, what the mother consumes is also consumed by the fetus or infant, with the exception of large organic ions such as heparin and insulin. In fact, virtually all (99%) drugs cross the placenta, and most medications penetrate human milk to some degree (Hale, 2012; Shephard & Lemire, 2007). Ideally, the pregnant or lactating woman should take as few drugs as possible, although exposure is usually more significant for the developing fetus than for the breastfeeding infant. Usually breastfeeding infants have less exposure because the concentrations of most medications in human milk are extraordinarily low, and with few exceptions, the dose delivered to the nursing infant is subclinical (Hale, 2012). Frequently, however, some form of drug therapy is necessary to treat the physiologic and hormonal changes that occur during pregnancy and postpartum, symptoms produced by the expanding uterus or functioning breast, and/or concomitant illnesses such as asthma, upper respiratory infection, epilepsy, or diabetes mellitus. Clinicians in the primary care setting should anticipate such complaints and should be prepared to treat patients appropriately. Unfortunately, despite the obvious need for drug therapy and the prevailing apprehension about using drugs in these special populations, clinicians may not be aware of information that is available to help them make astute clinical decisions. Because of the medicolegal implications of treating pregnant women and infants, pregnant women have been excluded from most clinical drug trials. Hence, evidence is insufficient regarding the safety and efficacy of many drug therapies during pregnancy. Notably, very few drugs have been granted FDA approval for use during pregnancy, and drugs that can be used during lactation are just being categorized. Thus, drugs for pregnant or lactating patients must be selected based on safety data derived from animal research and clinical data generated from case reports, retrospective case-control trials, and personal experience. What’s more, during lactation the care given to mothers may be confounded by breastfeeding misinformation. Consequently, to provide the most astute and contemporary care, those who treat the breastfed infant or the pregnant mother may have to consult the most accurate and up-to-date resources available. This includes using both telephone consultations and Internet resources for assistance in choosing the most appropriate medication (Box 6-1). Although the ideal condition may be the avoidance of any medications or chemicals during pregnancy, the FDA website reports that 90% of pregnant women have or develop medical problems that require them to take more than one prescription drug during pregnancy (Shephard & Lemire, 2007). According to its website, the average patient uses five to nine different drugs during pregnancy; 4% of pregnant women take more than 10 drugs during pregnancy; and 65% of women admit to self-administration of drugs during pregnancy. Additional research has demonstrated that the extent of fetal exposure to drugs may be vastly underestimated; patient medical charts reveal less than one fourth of the drugs actually consumed by the patient (Briggs, 2011; Koren, 2006; Gabbe et al, 2012). The desire for a healthy baby is universal. Mothers cannot help but worry about the health of their unborn child. Regardless of the mother’s concerns, many factors not under her control may influence the outcome of the pregnancy. Information about the thalidomide tragedy in the early 1960s, fueled by regulatory agencies, lawyers, and the public, has stimulated an intense search for the origin, prevention, and treatment of congenital malformations (Briggs, 2011). However, although vigilance in this area has been heightened, most knowledge about the effect of different drugs on the fetus comes from the experience and observation of clinicians—not from scientific research. Congenital malformations occur in many different forms, and their incidence varies widely. The incidence of major malformations in the general population is usually quoted as 2% to 3%, or 20 to 30 per 1000 live births. This translates into 200,000 birth defects annually (Schardein & Macina, 2006). Major malformations are those that are incompatible with survival (e.g., anencephaly) or that require major surgery for correction (e.g., cleft palate, congenital heart disease). If minor malformations are included (e.g., ear tags, extra digits), the rate of occurrence may be as high as 10% (Briggs, 2011). Congenital malformations account for about 14% of all infant deaths. Problems that arise during fetal development or within 1 month after birth account for two thirds of all infant mortality in this country (Gabbe et al, 2012; Schardein & Macina, 2006). Common lore blames drug exposure of the pregnant mother for any type of fetal damage that occurs. However, less than 5% of congenital malformations are probably related to drugs (Briggs, 2011). Only 19 drugs or groups of drugs have been identified as probable teratogenic agents in humans (Table 6-1). This contrasts with the almost 1000 teratogens that have been identified for laboratory animals (Schardein & Macina, 2006). This public misperception probably grew from media attention that surrounded the extreme fetal deformities that occurred as part of the thalidomide tragedy of the 1960s. TABLE 6-1 Drugs Considered to Be Teratogenic in Humans From Schardein JL, Macina OT: Human developmental toxicants: aspects of toxicology and chemistry, New York, 2006, CRC; Gabbe SG et al: Obstetrics—normal and problem pregnancies, ed 5, New York, 2012, Churchill Livingstone, Inc. Thalidomide is a central nervous system depressant that was used as a sedative-hypnotic agent and to reduce the nausea and vomiting of pregnancy. As many as 10,000 children were reportedly deformed as the result of use of this drug in as many as 30 countries, although the confirmed number is closer to 8000. The thalidomide episode focused the attention of both the scientific community and the lay community on the question of safe drug use in pregnant women. This drug was said to increase the rate of dysmelia by 80%, up to a rate of about 3:1000 to 5:1000 births. The reported malformations resulted when thalidomide was taken on days 21 to 36 after conception (days 34 to 50 postmenses). The risk that a woman will have a malformed child following thalidomide ingestion has been estimated to range from 2% to 25%, and retrospective analysis suggests that the mortality rate is 45%. Today, much controversy surrounds thalidomide research in women affected by human immunodeficiency virus and rheumatoid arthritis (Gabbe et al, 2012; Schardein & Macina, 2006; Shephard & Lemire, 2007). The most important determinant of the teratogenicity of an agent is the timing of the drug exposure (Figure 6-1). Exposure to drugs during the preimplantation phase results in an all-or-none effect: The affected cells either die or are described as undifferentiated, or totipotential, meaning that if one cell is damaged, another can assume its function (Briggs, 2011; Schardein & Macina, 2006). During this period, no malformations can be induced because there is no cell differentiation to allow any selective toxic reaction. Exposure to a teratogen may be lethal to the ovum, or the ovum may regenerate completely after exposure to a sublethal dose. Thus, exposure during this time may kill the conceptus, and the patient may not realize that she is pregnant; however, if the pregnancy continues, the risk of congenital anomalies is not increased (Briggs, 2011). The most critical period in which drug exposure should be avoided is the embryonic period (weeks 3 through 8), during which major organogenesis occurs and the risk of inducing major malformations is greatest. The damage induced by a drug administered during this period will depend on what organ systems were forming during the time of exposure. Because many organ systems form in parallel, multiple congenital defects may result from one drug exposure. The classic teratogenic period in humans lasts from 31 days after the last menstrual period through 10 weeks from the last menstrual period (Briggs, 2011), which corresponds to the period of organogenesis (14 to 56 days). After embryogenesis occurs, the organ structures continue to grow and mature physiologically during the fetal period (weeks 9 through term). During the fetal period (57 days to term), major malformations are not likely to occur, yet organ systems formed during the embryonic period may be damaged by exposure during the second or third trimester (Shephard & Lemire, 2007; Weiner & Buhimschi, 2003), and anomalies are more likely to involve functional aspects such as mental development and reproduction or fetal growth. Thus, exposure to a teratogen during this period may result in intrauterine growth retardation, and, because the central nervous system continues to develop throughout gestation, exposure may cause mental retardation or subtle, delayed behavioral effects. The placenta plays an important role in determining the teratogenic potential of a drug, primarily by allowing drugs to reach the fetus but perhaps also by allowing for the biotransformation of drugs before they reach the fetal circulation. The surface area of the placenta increases during gestation, while placental thickness decreases. Both of these structural changes favor the transfer of chemicals to the fetus (Briggs, 2011). In fact, as was stated earlier, nearly all drugs readily cross the placenta, reaching fetal concentrations of 50% to 100% of those in the maternal circulation. Primarily, the physiologic processes that govern the passage of drugs across the placenta are the same as those that apply to the passage of drugs across any lipid membrane. Once the drug has crossed the placenta, it is in the fetal circulation. Several other physicochemical properties affect the rate and/or extent of placental transfer, including lipid solubility, protein binding, and pH of the mother and fetus (Briggs, 2011). Currently, little is known about the contribution of the placenta to the metabolism of drugs that pass through it. However, metabolic inactivation of drugs by the placenta appears to be of less clinical concern than is the potential for the placenta to metabolize less active compounds to toxic metabolites (Koren, 2006). Several other factors that illustrate the principles of teratology include the following (Briggs, 2011; Gabbe et al, 2012; Schardein & Macina, 2006): • Maternal-fetal genotype—Maternal absorption, metabolism, and distribution; placental transfer; and fetal metabolism characteristics unique to each maternal-fetal pair as a result of genetic heterogeneity influence fetal susceptibility to a potential teratogen. This is easy to understand when clinicians observe that for the same teratogen, some individuals will prove especially susceptible, whereas others will be unusually resistant. • Dose-response relationships—The amount of medication taken often correlates with the observed response. Aberrant development may range from no effect at low doses to organ-specific malformations at intermediate doses to embryo-fetal toxicity at high doses. The extent of damage is also influenced by the stage of development and the route of administration. • Specificity of agent—The extent of adverse environmental influences on developing tissues depends heavily on the agent involved. Some agents have greater teratogenic potential than others, resulting in part from factors such as drug dosage, maternal metabolism, and placental transfer. • Drug interactions—Two teratogens administered separately may have a very different effect when given together. Induction or inhibition of enzyme systems and competition for binding sites caused by the two drugs may influence the levels of unbound and active teratogens. In general, animal models predict poorly whether a drug or chemical is a human teratogen, and it is extraordinarily difficult to directly extrapolate findings in animals to pregnant women (Gabbe et al, 2012). As clinicians gain experience with a drug, case reports may provide the first evidence that an agent is teratogenic in humans. Although human investigations are necessary to demonstrate that an agent is teratogenic, such studies are not informative until the agent has already damaged many children (Gabbe et al, 2012). For the best sources of information on potential teratogens, to report or investigate an exposure, or to obtain a list of current pregnancy exposure registries, see Box 6-1. The safe use of a drug in a single pregnancy or even in a large number of pregnancies does not ensure that the drug is safe in all pregnancies (Briggs, 2011). Very few drugs can be declared “safe in pregnancy.” The present state of knowledge does not allow prediction with any degree of certainty as to when a particular drug will prove teratogenic to a particular fetus. References can describe only relative risks for a specific population—not specific risks for specific patients (Briggs, 2011). To help quantify the measure of risk that a drug presents to the fetus, the FDA developed a classification scheme to aid in the selection of drug therapy for pregnant women, and all drugs must be labeled with an FDA pregnancy category rating (Table 6-2). These standard ranking of drugs for risk in pregnant and nursing women have been criticized as being confusing and oversimplistic and sometimes leading to errors. Beginning in 2008 the FDA drafted proposed rules change that would require drug manufacturer companies to comply with new product labeling guidelines. These guidelines would include data from pregnancy registries created to monitor response to drugs, as well as a concise narrative describing both risks and strategies to reduce unintentional exposures. The pregnancy labeling will replace the brief risk category labeling used now and include a clinical management statement, a summary risk assessment, and a discussion of data, with available human data presented before animal data (www.FDA.gov/, 2012). TABLE 6-2 From Meadows M: Pregnancy and the drug dilemma, FDA Consumer Magazine, 2001 (Available at www.fda.gov/fdac/features/2001/301_preg.html#categories). When a clinician discovers that a pregnant woman has been exposed to a dangerous drug, it is important to provide the patient with as much information as possible. First, determine whether the fetus was exposed during organogenesis; if so, refer the patient for a detailed ultrasonogram and to a perinatologist. If the exposure occurred outside organogenesis, then ordering an ultrasonogram to reassure the mother is an option. With drugs that have a high potential for fetal damage, the patient should be encouraged to make a thoughtful decision regarding whether to continue the pregnancy (Friedman & Polifka, 2007; Gabbe et al, 2012) (Box 6-2). Despite the many physiologic changes that occur during pregnancy that could theoretically affect absorption, bioavailability of the drug during pregnancy does not appear to be altered. Maternal blood volume increases by 30% to 40% (500 to 1800 ml) to support the requirements of the developing fetus. This may lead to decreased plasma concentrations of some drugs. Decreased albumin and α1-acid glycoprotein concentrations during pregnancy will result in decreased protein binding for highly bound drugs. For drugs metabolized by the liver, this can result in misinterpretation of total plasma concentrations of low–extraction ratio drugs and overdosing of high–extraction ratio drugs administered by nonoral routes. Renal clearance and the activity of the CYP isozymes—CYP 3A4, 2D6, and 2C9, and uridine 5’-diphosphate glucuronosyl transferase—are increased during pregnancy. In contrast, CYP 1A2 and 2C19 activity is decreased (Anderson, 2006). Renal function improves during gestation because renal plasma flow increases by 30% and the glomerular filtration rate (GFR) increases by as much as 50%. Because of this improved renal filtration, serum urea, creatinine, and uric acid levels usually are decreased in pregnancy. Cardiac output increases by as much as 32% because of an increased heart rate (up 10 to 15 bpm) and increased stroke volume (Hale, 2012). Not all changes in the system are positive to the mother. During pregnancy, a hypercoagulable state develops, with increased levels of fibrinogen and of factors VII, VIII, IX, and X. And, because bowel tone and gastrointestinal peristalsis decrease, constipation may become a problem for pregnant women. Pregnant women also have a high incidence of heartburn because of decreased gastrointestinal motility, increased estrogen and progesterone that decreases lower esophageal sphincter tone, and the increased pressure of the growing uterus on the abdomen (Hale, 2012). Nausea and vomiting, common symptoms during pregnancy, often are regarded as an unpleasant but normal part of pregnancy during the first and early second trimesters. Nausea and vomiting of pregnancy (NVP) occur in approximately 75% to 80% of pregnant women. NVP is self-limiting, typically starting 2 to 3 weeks after a missed menstrual period and continuing from the 8th to the 12th week of pregnancy. It is usually worse in the morning before the mother gets out of bed. The exact etiology and pathogenesis of NVP are poorly understood and are most likely multifactorial. Some theories for the etiology of NVP involve psychologic predisposition, evolutionary adaptation, hormonal stimuli, increased levels of human chorionic gonadotropin (hCG), and/or increased levels of progesterone associated with decreased gastric emptying, and Helicobacter pylori infection. Treatment ranges from dietary and lifestyle changes to vitamins, antiemetics, and hospitalization for intravenous therapy. Treatment generally begins with nonpharmacologic interventions; if symptoms do not improve, drug therapy is added. Although NVP has been associated with a positive pregnancy outcome, the symptoms can significantly affect a woman’s life, both personally and professionally. Given the substantial health care costs, as well as the indirect costs, and the potential decrease in quality of life due to NVP, providers must acknowledge the impact of NVP and provide appropriate treatment (Badell et al, 2006). Although NVP most frequently is self-limited, approximately 1% to 3% of pregnant women may experience severe nausea and vomiting, defined as hyperemesis gravidarum. This condition, which affects 3.5 of 1000 infants, is debilitating and can result in significant weight loss, electrolyte imbalance, ketosis, dehydration, and malnutrition. Overall, treatment of NVP should depend on the severity of symptoms, the impact of symptoms on a woman’s quality of life, and the safety of the fetus. Treatments range from dietary and lifestyle changes to vitamin supplementation, antiemetic therapy, and hospitalization. Treatment generally begins with nonpharmacologic interventions; drug therapy is added if nausea or vomiting does not improve. Such patients often require hospitalization for the administration of intravenous fluids and electrolytes, antiemetics, and sedation. Additionally, treatment with corticosteroids has been found to be effective (Gabbe et al, 2012). Benedictine (10 mg doxylamine/10 mg pyridoxine) was used for NVP in an estimated 10% to 25% of pregnant women in the United States from 1958 to 1983. In the 1960s, numerous birth defects (e.g., limb deformities, cleft palate, pyloric stenosis) associated with the use of Benedictine were reported worldwide (Schardein & Macina, 2006). However, the product has been studied since then, and those results have been refuted. Doxylamine and pyridoxine now carry a category B and A rating, respectively. Although the company voluntarily removed the branded product from the market in 1983, the active ingredients are available in other countries on a nonprescription basis (e.g., Unisom nighttime sleep aid, vitamin B6), or the product can be prescribed and compounded. A number of dopamine antagonists can be used for the treatment of NVP. During nausea and vomiting, dopamine receptors in the stomach mediate inhibition of gastric motility and may provide a site of action for antiemetic dopamine receptor antagonists. Dopamine, specifically at the dopamine2 (D2) receptors, is also implicated in emetic signaling through the chemoreceptor trigger zone. The three main classes of dopamine receptor antagonists are phenothiazines, butyrophenones, and benzamides. Low doses of phenothiazines antagonize the interaction of dopamine with D2 receptors to exert an antiemetic effect. Phenothiazines used in the treatment of NVP include prochlorperazine and promethazine. Metoclopramide, a benzamide, is a strong central and peripheral D2 antagonist. It exerts modest antiemetic effects by enhancing lower esophageal sphincter tone and decreasing transit time through the upper gastrointestinal tract (Badell et al, 2006). Corticosteroids have been evaluated for the treatment of severe NVP and hyperemesis gravidarum. Although a precise dosage has not been established for corticosteroids in the treatment of hyperemesis gravidarum, a possible regimen is oral or intravenous methylprednisolone 48 mg/day given in three divided doses for 2 to 3 days. If no response is seen within 3 days, the treatment should be stopped. If symptoms have not improved within 72 hours of the start of corticosteroid treatment, response beyond that time is not likely. Otherwise, the dosage may be tapered appropriately over 1 to 2 weeks. For women with recurrent vomiting, the tapered dosage may be stopped and the lowest effective dosage continued for up to 6 weeks. Corticosteroids should not be administered beyond this period for treatment of NVP because of maternal side effects (Badell et al, 2006). Table 6-3 contains a list of common antiemetics and other drugs used during pregnancy. TABLE 6-3 Recommended Drugs for Common Problems During Pregnancy From Gabbe SG et al: Obstetrics—normal and problem pregnancies, ed 5, New York, 2012, Churchill Livingstone, Inc.
Special Populations
Pregnant and Nursing Women
Drugs in Pregnant Women
Incidence
Principles of Teratology: Incidence and Types of Malformations
Drug
Major Defects
ACE inhibitors
Fetal renal dysplasia, fetal limb contractures, craniofacial deformities, hypoplastic lung development, fetal skull ossification defects
Antithyroid compounds, iodine
Hypothyroidism, goiter, scalp defects
Antibiotics
Aminoglycoside
Ototoxicity, eighth nerve
Tetracycline
Staining of teeth, hypoplasia enamel, inhibition of bone growth
Cephalosporins (cefaclor, cephalexin, cephradine)
Possible minor abnormalities
Anticancer agents
Polymorphic
methotrexate
Multiple congenital anomalies, cranial and malformation of extremities
Androgenic hormones
Masculinization, clitoromegaly, labial fusion
isotretinoin, etretinate
Structural anomalies 25%; mental retardation 25%; craniofacial, cardiac, thymic, CNS abnormalities; ear, mouth, and eye abnormalities; great vessel transposition of heart, ventricular septal defect
thalidomide
Severe limb, ear abnormalities
Antiepileptics
Double risk malformations: cleft lip, palate, cardiac
phenytoin
Fetal hydantoin syndrome: craniofacial, appendicular, cardiac, and skeletal, motor, and mental deficiency
lamotrigine
Congenital anomalies and seizure disorders
primidone
Microcephaly, cardiac, facial, mental deficiency
tegretol
Dysmorphic
valproic acid
Neural tube defect, spina bifida, facial defects
carbamazepine
Craniofacial, digital, neural tube defects
Coumarin anticoagulants
Nose, skeleton, CNS, ophthalmologic abnormalities, mental retardation
Alcohol
Fetal alcohol syndrome: facial, microcephaly, mental retardation, CNS dysfunction
Methadone
Facial, cardiac, urogenital, mental, and speech impairment
diethylstilbestrol (DES)
Uterine adenosis, cancer in females, accessory gonadal lesions in males
penicillamine
Skin hyperelasticity
Vitamin A analogs
Craniofacial, cardiac, CNS, thymic, mental retardation
Cocaine
Cardiovascular, CNS, neurologic defects
Thalidomide
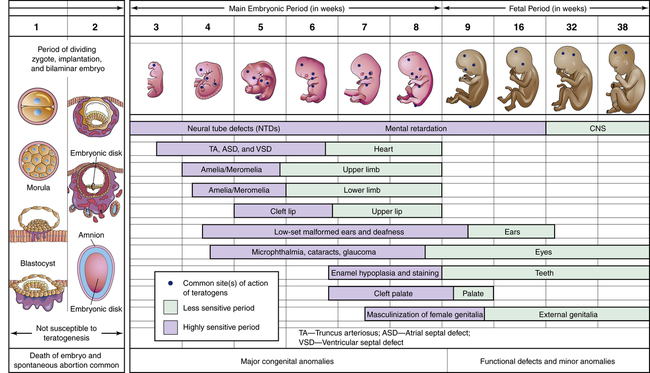
Determinants of Teratogenicity
Determining the Teratogenic Potential of Drugs
Counseling Pregnant Patients About Drug Use
Category
Description
A
Adequate, well-controlled studies in pregnant women have not shown an increased risk of fetal abnormalities. The possibility of fetal harm appears remote.
B
Animal studies have revealed no evidence of harm to the fetus; however, no adequate and well-controlled studies in pregnant women have been conducted.
Or Animal studies have shown an adverse effect, but adequate and well-controlled studies in pregnant women have failed to demonstrate a risk to the fetus.
C
Animal studies have shown an adverse effect, and no adequate and well-controlled studies in pregnant women have been conducted.
Or No animal studies have been conducted, and no adequate and well-controlled studies in pregnant women have been conducted. Give drugs only if the potential benefit justifies the potential risk to the fetus.
D
Studies, adequate well-controlled or observational, in pregnant women have demonstrated a risk to the fetus. However, the benefits of therapy may outweigh the potential risks. Give only if the drug is needed for a life-threatening situation or a serious disease for which safer drugs cannot be used or are ineffective.
X
Studies, adequate well-controlled or observational, in animals or pregnant women have demonstrated positive evidence of fetal abnormalities. Use of the product is contraindicated in women who are or may become pregnant.
Common Conditions Requiring Treatment During Pregnancy
Physiologic Changes
Nausea and Vomiting
Clinical Condition
Recommended Drugs
Nausea and vomiting
Antihistamines
dimenhydrinate (Dramamine) B
diphenhydramine (Benadryl) B in third trimester
meclizine (Antivert, Bonine) B
Phenothiazines
promethazine (Phenergan) C
prochlorperazine (Compazine) C
doxylamine C
metoclopramide (Reglan) B
phosphorated carbohydrate solution (Emetrol G)
pyridoxine (vitamin B6) A
Infections
Penicillins B
Cephalosporins B
(avoid cefaclor, cephalexin, cephradine)
Cardiovascular
Alpha-adrenergic receptor agonists
methyldopa (Aldomet)—B: Usually safe but benefits must outweigh the risks. Usually drug of choice.
labetalol (Normodyne, Trandate)—C: Safety for use during pregnancy has not been established.
pindolol (Visken)—B: Usually safe but benefits must outweigh the risks.
metoprolol (Lopressor, Toprol XL)—C: Safety for use during pregnancy has not been established.
Calcium channel blockers
nifedipine (Adalat, Procardia)—C: Safety for use during pregnancy has not been established.
Centrally acting alpha-adrenergic agonists
clonidine (Catapres)—C: Safety for use during pregnancy has not been established.
Diuretics
hydrochlorothiazide (Esidrix, HydroDIURIL [B])—D (expert analysis): Safety for use during pregnancy has not been established.
furosemide (Lasix)—C: Safety for use during pregnancy has not been established.
Vasodilators—decrease peripheral resistance by inducing vasodilation
nitroprusside (Nitropress)—C: Safety for use during pregnancy has not been established.
hydralazine (Apresoline)—C: Usually safe but benefits must outweigh the risks.
Anticonvulsant (for eclampsia)
Anticonvulsants—administered to prevent seizures in severe preeclampsia or eclampsia
phenytoin (Dilantin)—D: Unsafe but benefits may outweigh risks.
magnesium sulfate (Bilagog)—A: Safe in pregnancy.
Acne
Topical benzoyl peroxide—C
Topical clindamycin—B
Constipation
Bulk-forming laxatives (e.g., Metamucil, Citrucel, Perdiem)—C: Colace (docusate)—C: May be used later in pregnancy with less risk.
Heartburn/gastroesophageal reflux disease
MgAl combination antacids prn (Milk of Magnesia)
Lice
H2 antagonists ranitidine, cimetidine—B
Head lice
permethrin 1% cream rinse (Nix)—B
Pubic lice
permethrin 1% cream rinse (Nix) or pyrethrins with piperonyl butoxide—B
Scabies
permethrin 5% cream (Elimite)—B
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Special Populations: Pregnant and Nursing Women

Only gold members can continue reading. Log In or Register to continue
