59
Special Aspects of Perinatal & Pediatric Pharmacology
The effects of drugs on the fetus and newborn infant are based on the general principles set forth in Chapters 1–4 of this book. However, the physiologic contexts in which these pharmacologic laws operate are different in pregnant women and in rapidly maturing infants. At present, the special pharmacokinetic factors operative in these patients are beginning to be understood, whereas information regarding pharmacodynamic differences (eg, receptor characteristics and responses) is still incomplete.
DRUG THERAPY IN PREGNANCY
Pharmacokinetics
Most drugs taken by pregnant women can cross the placenta and expose the developing embryo and fetus to their pharmacologic and teratogenic effects. Critical factors affecting placental drug transfer and drug effects on the fetus include the following: (1) the physicochemical properties of the drug; (2) the rate at which the drug crosses the placenta and the amount of drug reaching the fetus; (3) the duration of exposure to the drug; (4) distribution characteristics in different fetal tissues; (5) the stage of placental and fetal development at the time of exposure to the drug; and (6) the effects of drugs used in combination.
A. Lipid Solubility
As is true also of other biologic membranes, drug passage across the placenta is dependent on lipid solubility and the degree of drug ionization. Lipophilic drugs tend to diffuse readily across the placenta and enter the fetal circulation. For example, thiopental, a drug commonly used for cesarean sections, crosses the placenta almost immediately and can produce sedation or apnea in the newborn infant. Highly ionized drugs such as succinylcholine and tubocurarine, also used for cesarean sections, cross the placenta slowly and achieve very low concentrations in the fetus. Impermeability of the placenta to polar compounds is relative rather than absolute. If high enough maternal-fetal concentration gradients are achieved, polar compounds cross the placenta in measurable amounts. Salicylate, which is almost completely ionized at physiologic pH, crosses the placenta rapidly. This occurs because the small amount of salicylate that is not ionized is highly lipid-soluble.
B. Molecular Size and pH
The molecular weight of the drug also influences the rate of transfer and the amount of drug transferred across the placenta. Drugs with molecular weights of 250–500 can cross the placenta easily, depending upon their lipid solubility and degree of ionization; those with molecular weights of 500–1000 cross the placenta with more difficulty; and those with molecular weights greater than 1000 cross very poorly. An important clinical application of this property is the choice of heparin as an anticoagulant in pregnant women. Because it is a very large (and polar) molecule, heparin is unable to cross the placenta. Unlike warfarin, which is teratogenic and should be avoided during the first trimester and even beyond (as the brain continues to develop), heparin may be safely given to pregnant women who need anticoagulation. Yet the placenta contains drug transporters, which can carry larger molecules to the fetus. For example, a variety of maternal antibodies cross the placenta and may cause fetal morbidity, as in Rh incompatibility.
Because maternal blood has a pH of 7.4 whereas the fetal blood is 7.3, basic drugs with a pKa above 7.4 will be more ionized in the fetal compartment, leading to ion trapping and, hence, to higher fetal levels (see Chapter 1, Ionization of Weak Acids and Weak Bases).
C. Placental Transporters
During the last decade, many drug transporters have been identified in the placenta, with increasing recognition of their effects on drug transfer to the fetus. For example, the P-glycoprotein transporter encoded by the MDR1 gene pumps back into the maternal circulation a variety of drugs, including cancer drugs (eg, vinblastine, doxorubicin) and other agents. Similarly, viral protease inhibitors, which are substrates to P-glycoprotein, achieve only low fetal concentrations—an effect that may increase the risk of vertical HIV infection from the mother to the fetus. The hypoglycemic drug glyburide has much lower plasma levels in the fetus as compared with the mother. Recent work has documented that this agent is effluxed from the fetal circulation by the BCRP transporter as well as by the MRP3 transporter located in the placental brush border membrane. In addition, very high maternal protein binding of glyburide (> 98.8%) also contributes to lower fetal levels as compared with maternal concentrations.
D. Protein Binding
The degree to which a drug is bound to plasma proteins (particularly albumin) may also affect the rate of transfer and the amount transferred. However, if a compound is very lipid-soluble (eg, some anesthetic gases), it will not be affected greatly by protein binding. Transfer of these more lipid-soluble drugs and their overall rates of equilibration are more dependent on (and proportionate to) placental blood flow. This is because very lipid-soluble drugs diffuse across placental membranes so rapidly that their overall rates of equilibration do not depend on the free drug concentrations becoming equal on both sides. If a drug is poorly lipid-soluble and is ionized, its transfer is slow and will probably be impeded by its binding to maternal plasma proteins. Differential protein binding is also important since some drugs exhibit greater protein binding in maternal plasma than in fetal plasma because of a lower binding affinity of fetal proteins. This has been shown for sulfonamides, barbiturates, phenytoin, and local anesthetic agents.
E. Placental and Fetal Drug Metabolism
Two mechanisms help protect the fetus from drugs in the maternal circulation: (1) The placenta itself plays a role both as a semipermeable barrier and as a site of metabolism of some drugs passing through it. Several different types of aromatic oxidation reactions (eg, hydroxylation, N-dealkylation, demethylation) have been shown to occur in placental tissue. Pentobarbital is oxidized in this way. Conversely, it is possible that the metabolic capacity of the placenta may lead to creation of toxic metabolites, and the placenta may therefore augment toxicity (eg, ethanol, benzpyrenes). (2) Drugs that have crossed the placenta enter the fetal circulation via the umbilical vein. About 40–60% of umbilical venous blood flow enters the fetal liver; the remainder bypasses the liver and enters the general fetal circulation. A drug that enters the liver may be partially metabolized there before it enters the fetal circulation. In addition, a large proportion of drug present in the umbilical artery (returning to the placenta) may be shunted through the placenta back to the umbilical vein and into the liver again. It should be noted that metabolites of some drugs may be more active than the parent compound and may affect the fetus adversely.
Pharmacodynamics
A. Maternal Drug Actions
The effects of drugs on the reproductive tissues (breast, uterus, etc) of the pregnant woman are sometimes altered by the endocrine environment appropriate for the stage of pregnancy. Drug effects on other maternal tissues (heart, lungs, kidneys, central nervous system, etc) are not changed significantly by pregnancy, although the physiologic context (cardiac output, renal blood flow, etc) may be altered, requiring the use of drugs that are not needed by the same woman when she is not pregnant. For example, cardiac glycosides and diuretics may be needed for heart failure precipitated by the increased cardiac workload of pregnancy, or insulin may be required for control of blood glucose in pregnancy-induced diabetes.
B. Therapeutic Drug Actions in the Fetus
Fetal therapeutics is an emerging area in perinatal pharmacology. This involves drug administration to the pregnant woman with the fetus as the target of the drug. At present, corticosteroids are used to stimulate fetal lung maturation when preterm birth is expected. Phenobarbital, when given to pregnant women near term, can induce fetal hepatic enzymes responsible for the glucuronidation of bilirubin, and the incidence of jaundice is lower in newborns when mothers are given phenobarbital than when phenobarbital is not used. Before phototherapy became the preferred mode of therapy for neonatal indirect hyperbilirubinemia, phenobarbital was used for this indication. Administration of phenobarbital to the mother was suggested recently as a means of decreasing the risk of intracranial bleeding in preterm infants. However, large randomized studies failed to confirm this effect. Antiarrhythmic drugs have also been given to mothers for treatment of fetal cardiac arrhythmias. Although their efficacy has not yet been established by controlled studies, digoxin, flecainide, procainamide, verapamil, and other antiarrhythmic agents have been shown to be effective in case series. During the last two decades it has been shown that maternal use of zidovudine decreases by two thirds transmission of HIV from the mother to the fetus, and use of combinations of three antiretroviral agents can eliminate fetal infection almost entirely (see Chapter 49).
C. Predictable Toxic Drug Actions in the Fetus
Chronic use of opioids by the mother may produce dependence in the fetus and newborn. This dependence may be manifested after delivery as a neonatal withdrawal syndrome. A less well understood fetal drug toxicity is caused by the use of angiotensin-converting enzyme inhibitors during pregnancy. These drugs can result in significant and irreversible renal damage in the fetus and are therefore contraindicated in pregnant women. Adverse effects may also be delayed, as in the case of female fetuses exposed to diethylstilbestrol, who may be at increased risk for adenocarcinoma of the vagina after puberty.
D. Teratogenic Drug Actions
A single intrauterine exposure to a drug can affect the fetal structures undergoing rapid development at the time of exposure. Thalidomide is an example of a drug that may profoundly affect the development of the limbs after only brief exposure. This exposure, however, must be at a critical time in the development of the limbs. The thalidomide phocomelia risk occurs during the fourth through the seventh weeks of gestation because it is during this time that the arms and legs develop (Figure 59–1).
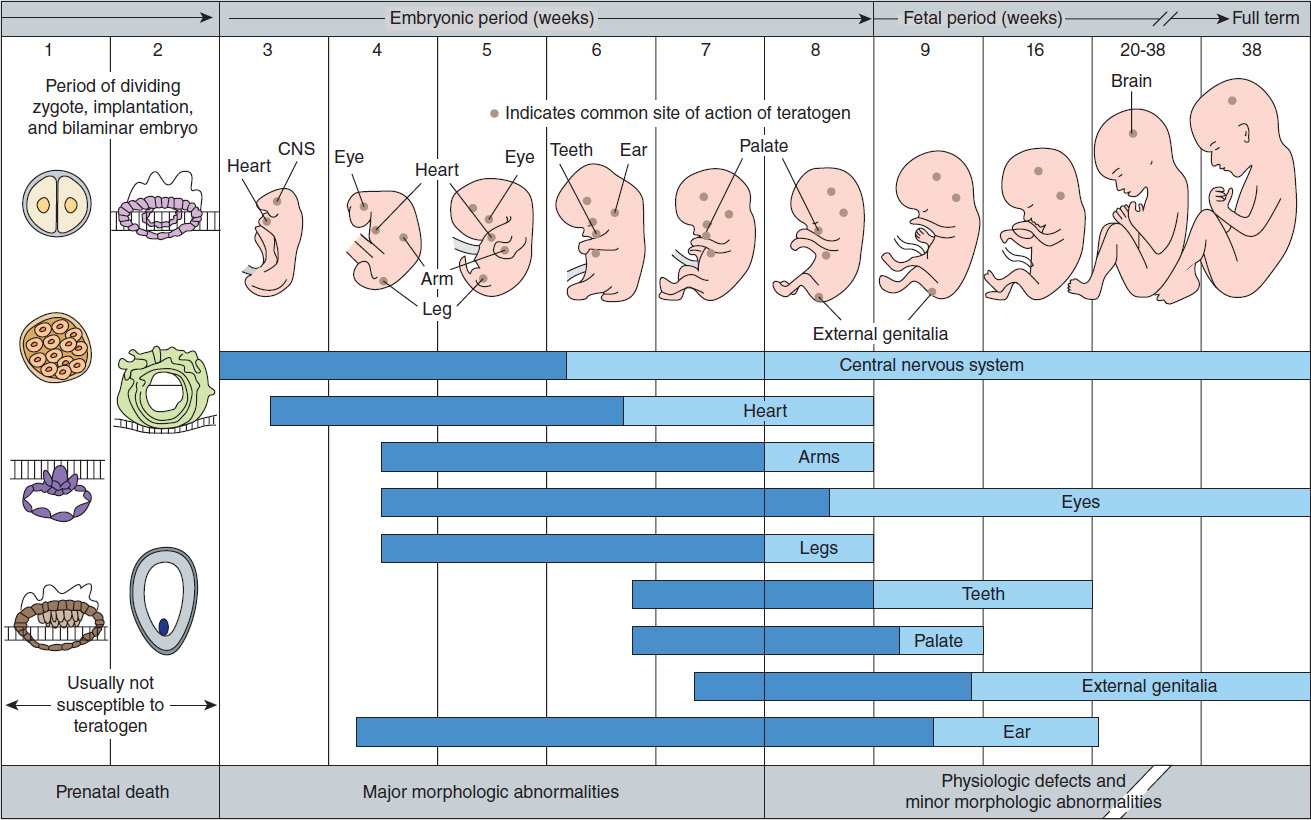
FIGURE 59–1 Schematic diagram of critical periods of human development. (Reproduced, with permission, from Moore KL: The Developing Human: Clinically Oriented Embryology, 4th ed. Saunders, 1988. © Elsevier.)
1. Teratogenic mechanisms—The mechanisms by which different drugs produce teratogenic effects are poorly understood and are probably multifactorial. For example, drugs may have a direct effect on maternal tissues with secondary or indirect effects on fetal tissues. Drugs may interfere with the passage of oxygen or nutrients through the placenta and therefore have effects on the most rapidly metabolizing tissues of the fetus. Finally, drugs may have important direct actions on the processes of differentiation in developing tissues. For example, vitamin A (retinol) has been shown to have important differentiation-directing actions in normal tissues. Several vitamin A analogs (isotretinoin, etretinate) are powerful teratogens, suggesting that they alter the normal processes of differentiation. Finally, deficiency of a critical substance appears to play a role in some types of abnormalities. For example, folic acid supplementation during pregnancy appears to reduce the incidence of neural tube defects (eg, spina bifida).
Continued exposure to a teratogen may produce cumulative effects or may affect several organs going through varying stages of development. Chronic consumption of high doses of ethanol during pregnancy, particularly during the first and second trimesters, may result in the fetal alcohol syndrome (see Chapter 23). In this syndrome, the central nervous system, growth, and facial development may be affected.
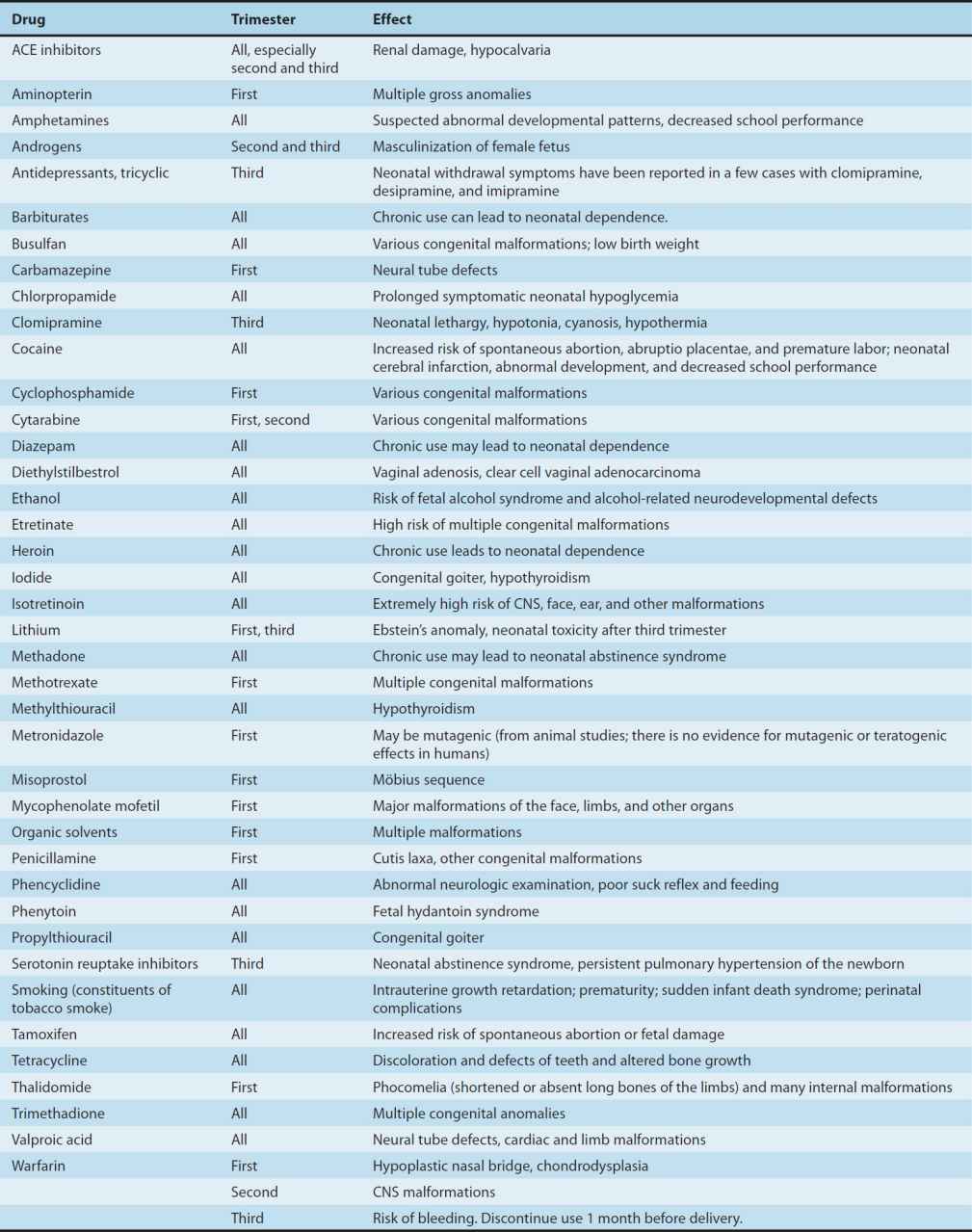
2. Defining a teratogen—To be considered teratogenic, a candidate substance or process should (1) result in a characteristic set of malformations, indicating selectivity for certain target organs; (2) exert its effects at a particular stage of fetal development, eg, during the limited time period of organogenesis of the target organs (Figure 59–1); and (3) show a dose-dependent incidence. Some drugs with known teratogenic or other adverse effects in pregnancy are listed in Table 59–1. Teratogenic effects are not limited only to major malformations, but also include intrauterine growth restriction (eg, cigarette smoking), miscarriage (eg, alcohol), stillbirth (eg, cigarette smoke), and neurocognitive delay (eg, alcohol, valproic acid).
TABLE 59–1 Drugs with significant teratogenic or other adverse effects on the fetus.
The widely cited FDA system for teratogenic potential (Table 59–2) is an attempt to quantify teratogenic risk from A (safe) to X (definite human teratogenic risk). This system has been criticized as inaccurate and impractical. For example, several drugs have been labeled “X” despite extensive opposite human safety data (eg, oral contraceptives). Diazepam and other benzodiazepines are labeled as “D” despite lack of positive evidence of human fetal risk. Presently the FDA is changing its system from the A, B, C grading system to narrative statements that will summarize evidence-based knowledge about each drug in terms of fetal risk and safety.
TABLE 59–2 FDA teratogenic risk categories.

3. Counseling women about teratogenic risk
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


