Solutions and mixtures: Nonelectrolytes
In Chapter 3, we began a discussion of mixtures and solutions of fluids. Many of the points we develop below will be applicable to mixtures in any phase. However, our emphasis here will be on liquid-phase mixtures of nonelectrolytes. In the treatment of equilibria we have seen that activities – not concentrations – are the fundamental quantities that are required to calculate the Gibbs energy (and therefore equilibrium constants, enthalpy, entropy and most other properties). That is especially true in the liquid phase because molecules are in close contact and it is impossible to ignore intermolecular interactions. In this chapter we will study in much more depth the concepts of activity, activity coefficients, and how they are measured. But let us begin with a definition of the ideal case. Note that we will concentrate on the simplest type of mixture, a binary mixture. A binary system has two components, a unary system has one component, a ternary has three components, a quaternary has four components, and so forth.
We will investigate the dependence of mixing parameters for a wide range of systems on a number of variables. Which variables are most important will depend on the system. Temperature and composition are two of the most important and they must always be specified. For liquids mixing with liquids, the pressure dependence of solubilities is negligible up to at least 100 atm or so. For gas/liquid solutions on the other hand, the pressure – or, more accurately, the partial pressure – of the gas is one of the most important parameters for determining the solubility. However, as we approach the critical point where the line between liquid and gas blurs and eventually disappears completely, pressure dependence becomes quite important.
13.1 Ideal solution and the standard state
Recall the definition of an ideal gas:
- No intermolecular interactions.
- Molecules have no volume.
- All gases obey the same equation of state: pV = nRT.
Note the similarities and difference to the properties of an ideal solution (in any phase):
- Intermolecular interactions are the same for all molecules, (Δmix H°)p, T = 0.
- Molecules have a finite volume independent of composition, (Δmix V)p, T = 0.
- The solvent follows Raoult’s law and the solute follows Henry’s law.
- The activity coefficient for all species is unity, that is, γ = 1.
- All mixtures are miscible (mix at all concentrations) for all temperatures.
- Mixing is driven only by a positive entropy change upon mixing, Δmix S° > 0.
We will explore the implications of this model and deviations from it throughout this chapter. As we shall see below, all solutions act as ideal dilute solutions when dilute enough (which may be very dilute indeed for solutions with dissimilar intermolecular interactions) but, in general, solutions in the liquid phase are non-ideal. Non-ideal solutions are called real solutions.
It is always important to construct an appropriate standard state. For a gas, we have chosen a standard state pressure of 1 bar or in SI units, p° = 100 kPa. Ideal solutions are miscible; that is, they form mixtures in which the composition may be varied from a mole fraction of 0 to 1 for both components. Therefore, we construct the standard state as the pure liquid. For simplicity, let us concentrate on binary solutions. Neither component can truly be considered a solute or solvent since the roles are reversed on either side of x = 0.5, where x is the mole fraction. Thus, in a binary mixture of components 1 and 2, the standard state mole fraction of component 1 is x°1 = 1, and similarly for component 2. For solids dissolved in a liquid, it will make more sense to set the standard state equal to a molality of b° = 1 mol kg− 1 or a molar concentration of c° = 1 mol l− 1. We will discuss the implications of changing the choice of standard state in the next chapter. Below, we will simplify our notation when using mole fraction. That is, there is no need to divide through by the standard state mole fraction of 1 since not only is this value unity, it is also already dimensionless.
13.2 Partial molar volume
In ideal solutions, volumes are additive. It may surprise you that for most real solutions this is not the case. That is, adding 50 ml of one liquid to 50 ml of another does not lead to a mixture of 100 ml final volume. To account for this we introduce the concept of partial molar volume. The partial molar volume is the change in volume upon addition of component 2 to component 1, holding all other variables – that is, p, T and moles of 1 – constant. The partial molar volume is not constant but depends on the solution composition. Taking V as the volume of the mixture, we write the partial molar volume of component j as

Equation (13.1) reflects the recommended IUPAC notation for partial molar volume. However, this notation is absolutely treacherous, as it implies the use of the same symbol for the partial molar volume of j and the volume of j. Hence, we adopt the notation of Berry, Rice and Ross and follow their derivations closely. In general, a partial molar quantity is defined as

Using this notation and definition to denote the partial molar volume of component j as  , we write the total change in volume for infinitesimal changes in molar composition of a binary mixture of 1 and 2 as
, we write the total change in volume for infinitesimal changes in molar composition of a binary mixture of 1 and 2 as

Thus,

The partial molar volume of component 1 depends on the composition of solution to which it is added because the effective volume of a given number of component 1 molecules depends on the identity of the molecules that surround them. As the concentration changes, the intermolecular forces acting on the molecules change. Therefore, the properties of real solutions and their components change as functions of mole fractions and chemical identities of the components. Molar volume is always positive; however, partial molar quantities need not be – that is, the addition of more liquid can lead to a contraction of the total volume. This is often true for small additions of a salt to an aqueous solution because the resulting ions must be strongly solvated. Even nonelectrolyte solutions – especially strongly interacting, hydrogen-bonding components – can exhibit complex partial molar volume changes with composition.
13.3 Partial molar Gibbs energy = chemical potential
13.3.1 Chemical potential of a one-component system
The molar Gibbs energy Gm and the chemical potential μ are synonymous for a one-component system. Thus, for any pure substance

We substitute for Gm in the fundamental equation and express this in differential form as

Since these are exact differentials, we can express how the chemical potential changes with temperature at constant pressure (dp = 0) according to the partial derivative

and how the chemical potential changes with pressure at constant temperature (dT = 0) according to the partial derivative

Since the molar entropy and molar volume of a gas are much greater than for the other two phases, it is clear from Eqs. (13.7) and (13.8) that the chemical potential of a gas responds to changes of temperature and pressure more sensitively than for liquids and solids. The importance of this can be observed directly in a plot of chemical potential versus temperature, as shown in Fig. 13.1. The partial derivative represents the slope of the curve. Each phase has its own characteristic dependence of molar entropy on temperature. Nonetheless, we know that the molar entropy of a gas is greater than that of a liquid at the same temperature. Similarly, the molar entropy of a liquid is always greater than that of a solid of the same material at the same temperature. Stated mathematically, at any given temperature Sm, g > Sm,l > Sm, s.

Figure 13.1 μ–T phase diagram for a material with properties approximately those of water. The chemical potential versus temperature of system at equilibrium is described by the curve with the lowest chemical potential.
The molar entropy of each phase is a function of temperature. In accord with Eq. (13.7) the chemical potential versus temperature of each phase is represented by a curve, as shown in Fig. 13.1. A system is at equilibrium when its chemical potential is a minimum. The lowest chemical potential phase is the most stable at any given temperature and pressure. Figure 13.1 represents the chemical potential versus temperature at the standard pressure of p° = 1 bar. The curve that represents the chemical potential of the solid starts with a slope of zero at absolute zero. All perfect crystals must behave this way according to the Third Law of Thermodynamics. The slope gradually becomes more negative. At the standard melting point Tf, the chemical potentials of the solid and liquid phases are equal, μ(s) = μ(l). At this point the curve for the chemical potential of the solid intersects the curve for the chemical potential of the liquid. We do not need to know the absolute value of either slope; however, we do know that the slope of the liquid curve is greater than the slope of the solid curve. Similarly, at the standard boiling point Tb, the chemical potentials of the liquid and gas phases are equal, μ(l) = μ(g). At this point the curve for the chemical potential of the liquid intersects the curve for the chemical potential of the gas, and the slope of the gas curve is greater than the slope of the liquid curve. In the previous chapter we developed the implications of these curves in more detail as we discussed the nature of phase transitions. Here, we focus on mixtures of different chemical components.
13.3.2 Chemical potential of a multicomponent system
Partial molar quantities can be defined using Eq. (13.2) for all thermodynamic properties of a mixture. One of particular importance is the partial molar Gibbs energy, which is equivalent to the chemical potential of that species. In a mixture with several components, the chemical potential of component j is

The chemical potential allows us to calculate the total Gibbs energy of an arbitrary mixture with r components

By definition, the Gibbs energy is related to internal energy, entropy, T, p and V by

For an infinitesimal change in composition of the mixture, Eq. (13.10) implies

whereas Eq. (13.11) demands

Equating these last two equations and solving for dU yields

However, the internal energy changes as

Substituting into Eq. (13.14) for dU and cancelling like terms of both sides gives

This is known as the Gibbs–Duhem equation. Therefore for constant p and T, the Gibbs–Duhem equation simplifies to

or more specifically for a binary solution

The Gibbs–Duhem equation shows us that the intensive variables T, p and μj are coupled. It places limits on their variability. At constant p and T, the chemical potentials of the components of a binary mixture cannot change independently of one another, only in concert, according to

Just as we saw for Eq. (4.23), because the summation in Eq. (13.17) is equal to a constant, there are r – 1 independent terms.
It follows from Eq. (13.15) and the usual definitions that



Let us reflect on these last three equations to obtain a better perspective of the fundamental importance of the chemical potential. Equations (13.20) to (13.22) tell us that the changes in enthalpy H, Helmholtz energy A, and Gibbs energy G as a function of composition are related to the chemical potential according to

Thus, using the general definition of partial molar quantities in Eq. (13.2), we can also write the chemical potential in terms of the fundamental relationship of H and S

where  are the partial molar enthalpy and partial molar entropy, respectively. We can now investigate the equilibrium properties of multicomponent systems.
are the partial molar enthalpy and partial molar entropy, respectively. We can now investigate the equilibrium properties of multicomponent systems.
13.4 The chemical potential of a mixture and ΔmixG
Equation (13.19) shows us that the addition of n1 moles of component 1 to a binary mixture requires that

Since both n1 and n2 are positive numbers, the derivatives must have opposite signs. The change in Gibbs energy upon addition of n1 moles is positive ΔG > 0; thus, the derivatives have signs consistent with


Experimentally, it is found that the right-hand side of Eq. (13.25) approaches a constant nonzero value represented by –B. Therefore, we substitute this value into Eq. (13.25) to obtain

Integration yields

Here, the integration ‘constant’ is constant under the chosen conditions of constant p, T and moles of component 2. However, in general, it is allowed to be a function of these variables. To set the value of this constant, recall that the chemical potential is an intensive property that cannot depend on the amount of 1 or 2 but only on their ratio. Furthermore, the chemical potential of the pure solution must correspond to μ°(p,T), the standard state chemical potential at the chosen values of p and T. This must be so because we have chosen the standard state to be the same as the pure substance. That is, if we introduce the mole fraction x1 = n1/(n1 + n2), Eq. (13.29) must behave as

It must also have a limiting behavior of the form

Therefore, the chemical potential as a function of composition is given by

Now it remains to set the value of the constant B. The Gibbs energy of mixing is obtained by the difference of the final mixed state Gibbs energy with the Gibbs energy of the initially unmixed state

From Eq. (13.6), we write for the Gibbs energy of mixing

Here, we consider the mixing of an ideal solution at constant p and T. Recall that in an ideal solution neither the enthalpy nor volume change upon mixing. Thus, with the aid of the thermodynamic definition of these two variables, we have


and

Equations (13.35)–(13.37) require that the difference of chemical potentials has the form

where the functions fj(n1, …, nr) are functions of composition alone. Looking at Eq. (13.37) and substituting from Eqs. (13.34) and (13.18), we obtain

This equation for the entropy of mixing is completely general. We have not assumed any particular phase, only that the solution behaves ideally. Because the functions fj(n1, …, nr) do not depend on p and T, the equation must be valid for all ideal solutions, regardless of the values of p and T. At sufficiently high T and low p, the system will behave in the ideal gas limit. As we will confirm below, and have shown before, the entropy of mixing of ideal gases is

Therefore, Eqs. (13.39) and (13.40) can only be consistent if

which means that the chemical potential of a component in an ideal solution as a function of composition is given by

13.5 Activity
In the most general terms, Gibbs energy is related to activity. The activity a is related to concentration by the activity coefficient γ. Activity can be thought of as the effective concentration of a species. The corresponding activity coefficient is a measure of how close to ideal that species is acting. The type of concentration term that we use depends on the system at hand. It is related to the choice of standard state. In this example, and much of this chapter, we are going to use concentration in terms of mole fraction x. Take A to be the solvent and B the solute:

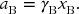
The solute and solvent each have different values for activity coefficients. Since we have a binary solution, xA + xB = 1. The chemical potential of a liquid A (the solvent in this case) is related to the activity by

As stated above, the standard state is chosen to be the pure substance. Hence, μ*A(l) is the chemical potential of the pure liquid. Substituting from Eq. (13.43) for the activity, we find that the chemical potential of any real solution is
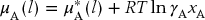
Real Solution:

By making the identification that in an ideal solution

and therefore

then the chemical potential of the solvent in an ideal solution is given by
Ideal Solution:

This, of course, corresponds to what we derived in Eq. (13.42). The deviation from ideal behavior is accounted for by the RT ln γA term in Eq. (13.47). The singly underlined terms represent the ideal contribution to the chemical potential.
All solutions behave more ideally as they become more dilute, that is aA → xA as xA → 1. Since xA < 1, ln xA < 0. In other words, according to Eq. (13.50), the presence of the solute stabilizes the solvent in a solution compared to the pure solvent.
By analogy with the above for solvents, we perform a similar calculation for the solute

The value of μ°B(l) depends on the standard state we chose for the solute. If instead of mole fraction, we work with molality, then we assume a standard state at molality b° = 1 mol kg–1, such that

Ideal behavior of the solute, as defined by γB → 1, is observed when the solute is very dilute, or equivalently as bB → 0. The choice of a standard state in terms of molality or molarity is common for electrolytes in solution. It may also be used for immiscible nonelectrolytes. When the standard state is chosen in this manner, we say that the activity coefficient is referenced to Henry’s law. As we shall see below, Henry’s law describes the behavior we expect of a solute in solution. Specifically, it describes the partial pressure of a volatile solute above a solution.
For miscible nonelectrolytes, it is more common to take the standard state to be the pure substance. In this case the activity coefficient of B approaches 1 when the solution approaches pure B. When the activity coefficient is chosen to be referenced in this manner, we say that the activity coefficient is referenced to Raoult’s law. As we shall see below, Raoult’s law defines the behavior we expect of the vapor pressure of the majority component above a solution. IUPAC now recommends the use of the symbol f rather than γ when this standard state is chosen.
Officially, while the activity is equal to the product of activity coefficient and concentration, it is defined in terms of the chemical potential,

The term relative activity is sometimes also used to differentiate aB from the absolute activity  .
.
13.5.1 Fugacity
Deviations from ideal behavior of real gases, which arise from intermolecular forces and finite molecular volume, are dealt with by introducing the concept of fugacity. Fugacity is analogous to activity. It is represented by either f or  , and is related to partial pressure according to
, and is related to partial pressure according to

φ is the fugacity coefficient. It depends on the identity of the gas, p and T. Unlike activity, which is dimensionless, fugacity has the same units as pressure. All gases behave more ideally as the pressure drops, that is φ → 1 as p → 0. Formally, we can define the fugacity of component B with partial pressure pB according to

where Vm is the molar volume and λB is the absolute activity. Substituting into the equation for the chemical potential of a gas, we obtain for a real gas


Note the similarity to the expression derived for solvent activity. The chemical potential can be written as the sum of an ideal term (single underline) and a term involving the fugacity coefficient that embodies the deviations from ideal behavior (double underline).
13.6 Measurement of activity
At equilibrium the chemical potential must be the same everywhere in the system, as shown in Fig. 13.2. Therefore, the chemical potentials of two phases in contact must be equal, and for a liquid in contact with its vapor


Figure 13.2 When a liquid solution equilibrates with the vapor phase, the concentrations (here in terms of the mole fractions of the two components in solution xA and xB as opposed to yA and yB in the gas phase) adjust until the chemical potential of every species in every phase is the same. The partial pressures of A and B sum to the total pressure, ptotal = pA + pB.
The * indicates a pure substance. We know how to write the chemical potential for the ideal gas phase

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


