CHAPTER 12 Skeletal Muscle Physiology
Muscle cells are highly specialized cells for the conversion of chemical energy to mechanical energy. Specifically, muscle cells use the energy in ATP to generate force or do work. Because work can take many forms (such as locomotion, pumping blood, or peristalsis), several types of muscle have evolved. The three basic types of muscle are skeletal muscle, cardiac muscle, and smooth muscle.
ORGANIZATION OF SKELETAL MUSCLE
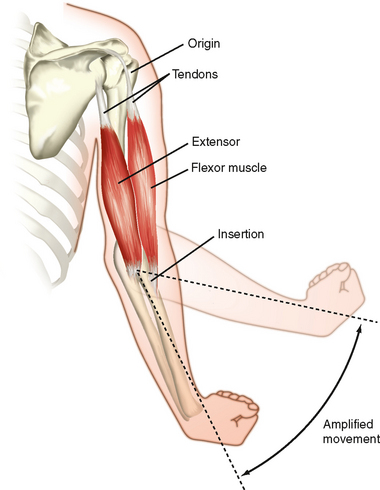
Figure 12-1 illustrates skeletal muscles spanning the elbow joint. The muscles are attached to bone on either side of the joint. The point of attachment closest to the spine is called the origin, whereas the point of attachment on the distal region (on the far side of the joint) is called the insertion. These points of attachment occur through tendons (connective tissue) at the end of the muscle. Note that the point of insertion is close to the elbow joint, which promotes a broad range of motion. Also note that the joint is spanned by a flexor muscle on one side and an extensor muscle on the opposite side of the joint. Thus, contraction of the flexor muscle (see the biceps muscle in Fig. 12-1) results in a decrease in the angle of the elbow joint (bringing the forearm closer to the shoulder), whereas contraction of the extensor muscle (see the triceps muscle in Fig. 12-1) results in the reverse motion (extending the arm).
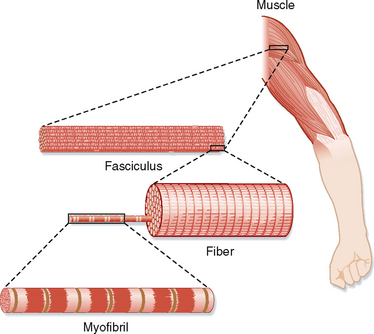
The basic structure of skeletal muscle is shown in Figure 12-2. Each muscle is composed of numerous cells called muscle fibers. A connective tissue layer called the endomysium surrounds each of these fibers. Individual muscle fibers are then grouped together into fascicles, which are surrounded by another connective tissue layer called the perimysium. Within the perimysium are the blood vessels and nerves that supply the individual muscle fibers. Finally, fascicles are joined together to form the muscle. The connective tissue sheath that surrounds the muscle is called the epimysium. At the ends of the muscle, the connective tissue layers come together to form a tendon, which attaches the muscle to the skeleton. The connective tissue layers are composed mainly of elastin and collagen fibers, and they serve to transmit movement of the actin and myosin molecules to the skeleton to effect movement. The connective tissue layers also contribute to passive tension of muscle and prevent damage to the muscle fibers as a result of overstretching or contraction (or both).
A myofibril can be subdivided longitudinally into sarcomeres (Fig. 12-3). The sarcomere is demarcated by two dark lines called Z lines and represents a repeating contractile unit in skeletal muscle. The average length of a sarcomere is 2 μm. On either side of the Z line is a light band (I band) that contains thin filaments composed primarily of the protein actin. The area between two I bands within a sarcomere is the A band, which contains thick filaments composed primarily of the protein myosin. The thin actin filaments extend from the Z line toward the center of the sarcomere and overlap a portion of the thick filaments. The dark area at the end of the A band represents this region of overlap between thick and thin filaments. A light area present in the center of the sarcomere is called the H band. This area represents the portion of the A band that contains myosin thick filaments, but no thin actin filaments. Thus, thin actin filaments extend from the Z line to the edge of the H band and overlap a portion of the thick filament in the A band. A dark line called the M line is evident in the center of the sarcomere and includes proteins that appear to be critical for organization and alignment of the thick filaments in the sarcomere.
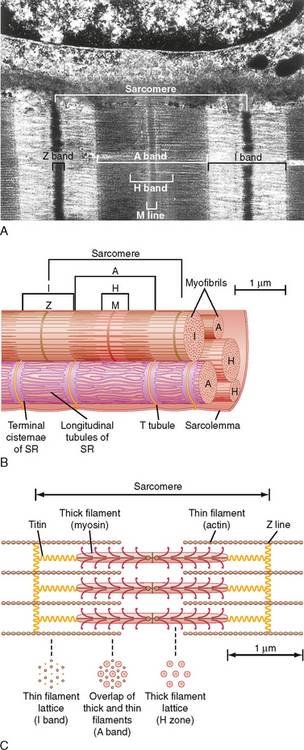
As illustrated in Figure 12-3, each myofibril in a muscle fiber is surrounded by sarcoplasmic reticulum (SR). The SR is an intracellular membrane network that plays a critical role in the regulation of intracellular [Ca++]. Invaginations of the sarcolemma, called T tubules, pass into the muscle fiber near the ends of the A band (i.e., close to the SR). The SR and the T tubules, however, are distinct membrane systems. The SR is an intracellular network, whereas the T tubules are in contact with the extracellular space. A gap (≈15 nm in width) separates the T tubules from the SR. The portion of the SR nearest the T tubules is called the terminal cisternae, and it is the site of Ca++ release, which is critical for contraction of skeletal muscle (see later). The longitudinal portions of the SR are continuous with the terminal cisternae and extend along the length of the sarcomere. This portion of the SR contains a high density of Ca++ pump protein (i.e., Ca++-ATPase), which is critical for reaccumulation of Ca++ in the SR and hence relaxation of the muscle.
The thick and thin filaments are highly organized in the sarcomere of myofibrils (Fig. 12-3). As mentioned, thin actin filaments extend from the Z line toward the center of the sarcomere, whereas thick myosin filaments are centrally located and overlap a portion of the opposing thin actin filaments. The thick and thin filaments are oriented such that in the region of overlap within the sarcomere, each thick myosin filament is surrounded by a hexagonal array of thin actin filaments. It is the Ca++-dependent interaction of the thick myosin and the thin actin filaments that generates the force of contraction after stimulation of the muscle (see later).
The thick myosin filaments are tethered to the Z lines by a cytoskeletal protein called titin. Titin is a very large elastic protein (molecular weight in excess of 3000 kDa) that extends from the Z line to the center of the sarcomere and appears to be important for organization and alignment of the thick filaments in the sarcomere. Titin may also serve as a mechanosensor and influence gene expression and protein degradation in a mechanical activity–dependent manner. Some forms of muscular dystrophy have been attributed to defects in titin.
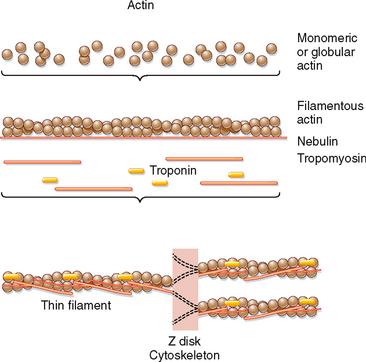
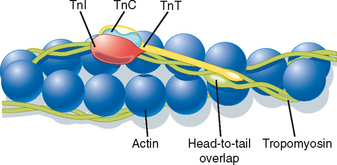
The thin filament is formed by the aggregation of actin molecules (termed globular actin or G-actin) into a two-stranded helical filament called F-actin, or filamentous actin (Fig. 12-5). The elongated cytoskeletal protein nebulin extends along the length of the thin filament and may participate in regulation of the length of the thin filament. Dimers of the protein tropomyosin extend over the entire actin filament and cover myosin binding sites on the actin molecules. Each tropomyosin dimer extends over seven actin molecules, with sequential tropomyosin dimers arranged in a head-to-tail configuration. A troponin complex consisting of three subunits (troponin T, troponin I, and troponin C) is present on each tropomyosin dimer and influences the position of the tropomyosin molecule on the actin filament and hence the ability of tropomyosin to inhibit binding of myosin to the actin filament. Troponin T binds tropomyosin, troponin I facilitates the inhibition of myosin binding to actin by tropomyosin, and troponin C binds Ca++. Binding of Ca++ to troponin C promotes the movement of tropomyosin on the actin filament, thereby exposing myosin binding sites and facilitating the interaction of myosin and actin filaments and sarcomere contraction (see later). Additional proteins associated with the thin filament include tropomodulin, α-actinin, and capZ protein. Tropomodulin is located at the end of the thin filament, toward the center of the sarcomere, and may participate in setting the length of the thin filament. α-Actinin and capZ protein serve to anchor the thin filament to the Z line.
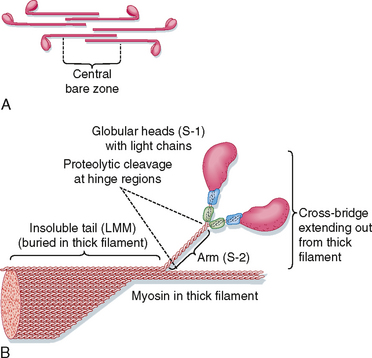
Organization of the thick filament is shown in Figure 12-6. Myosin is a large protein (≈480 kDa) that consists of six different polypeptides with one pair of large heavy chains (≈200 kDa) and two pairs of light chains (≈20 kDa). The heavy chains are wound together in an α-helical configuration to form a long rod-like segment, with the N-terminal portion of each heavy chain forming a large globular head. The head region extends away from the thick filament toward the actin thin filament and is the portion of the molecule that can bind to actin. Myosin is also able to hydrolyze ATP, and ATPase activity is located in the globular head as well. The two pairs of light chains are associated with the globular head. One of these pairs of light chains, termed essential light chains, is critical for the ATPase activity of myosin. The other pair of light chains, sometimes called regulatory light chains, may influence the kinetics of myosin and actin binding under certain conditions. Thus, myosin ATPase activity resides in the globular head of myosin and requires the presence of light chains (viz., the “essential” light chains).
The muscular dystrophies constitute a group of genetically determined degenerative disorders. Duchenneapos;s muscular dystrophy (DMD; described by G.B. Duchenne in 1861) is the most common of the muscular dystrophies and affects 1 in 3500 boys (3 to 5 years of age). Severe muscle wasting occurs, with most patients being wheelchair bound by the age of 12 and many dying of respiratory failure in adulthood (30 to 40 years of age). DMD is an X-linked recessive disease that has been linked to a defect in the dystrophin gene that leads to a deficiency of the dystrophin protein in skeletal muscle, brain, retina, and smooth muscle. Dystrophin is a large (427 kDa) protein that is present in low abundance (0.025%) in skeletal muscle. It is localized on the intracellular surface of the sarcolemma in association with several integral membrane glycoproteins (forming a dystrophin-glycoprotein complex). This dystrophin-glycoprotein complex provides a structural link between the subsarcolemmal cytoskeleton of the muscle cell and the extracellular matrix (Fig. 12-4) and appears to stabilize the sarcolemma and hence prevents contraction-induced injury (rupture). The dystrophin-glycoprotein complex may also serve as a scaffold for cell signaling cascades that promote cell survival.
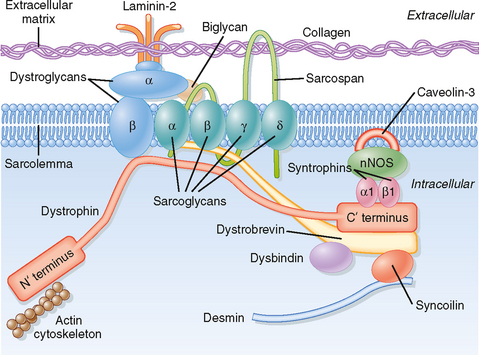
Although defects in the dystrophin-glycoprotein complex are involved in many forms of muscular dystrophy, recent studies have identified some forms of muscular dystrophy that involve other mechanisms. Specifically, a defect in sarcolemma repair (attributed to loss/mutation of the protein dysferlin) appears to underlie at least one form of muscular dystrophy (limb-girdle muscular dystrophy 2B, associated with muscle wasting in the pelvic region). Defects in the protein titin (termed titinopathies) have been implicated in other forms of muscular dystrophy (e.g., limb-girdle muscular dystrophy 2J and tibial muscular dystrophy). The link between titin mutations and muscular dystrophy may reflect a disruption in the ability of titin to bind a signalosome that can inhibit transcription and promote protein degradation. In the latter mechanism, the signalosome has been shown to include a muscle-specific ubiquitin ligase (viz., MuRF2) that can inhibit a transcription factor (viz., serum response factor) by promoting translocation to the cytosol and promote protein degradation (through ubiquitinationapos;see Chapter 1). Mutations in the protease calpain 3 (resulting in loss of protease activity) have also been implicated in some types of muscular dystrophy (e.g., limb-girdle muscular dystrophy 2A), apparently secondary to apoptosis.
Myosin filaments form by a tail-to-tail association of myosin molecules, thereby resulting in a bipolar arrangement of the thick filament. The thick filament then extends on either side of the central bare zone by a head-to-tail association of myosin molecules, thus maintaining the bipolar organization of the thick filament centered on the M line. Such a bipolar arrangement is critical for drawing the Z lines together (i.e., shortening the length of the sarcomere) during contraction. The mechanisms controlling this highly organized structure of the myosin thick filament are not clear, although the cytoskeletal protein titin is thought to participate in the formation of a scaffold for organization and alignment of the thick filament in the sarcomere. Additional proteins found in the thick filaments (e.g., myomesin and C protein) may also participate in the bipolar organization or packing of the thick filament (or both).
CONTROL OF SKELETAL MUSCLE ACTIVITY
Motor Nerves and Motor Units
Skeletal muscle is controlled by the central nervous system. Specifically, each skeletal muscle is innervated by an α motor neuron. The cell bodies of α motor neurons are located in the ventral horn of the spinal cord (Fig. 12-7; see also Chapter 9). The motor axons exit via the ventral roots and reach the muscle through mixed peripheral nerves. The motor nerves branch in the muscle, with each branch innervating a single muscle fiber. The specialized cholinergic synapse that forms the neuromuscular junction and the neuromuscular transmission process that generates an action potential in the muscle fiber are described in Chapter 6.
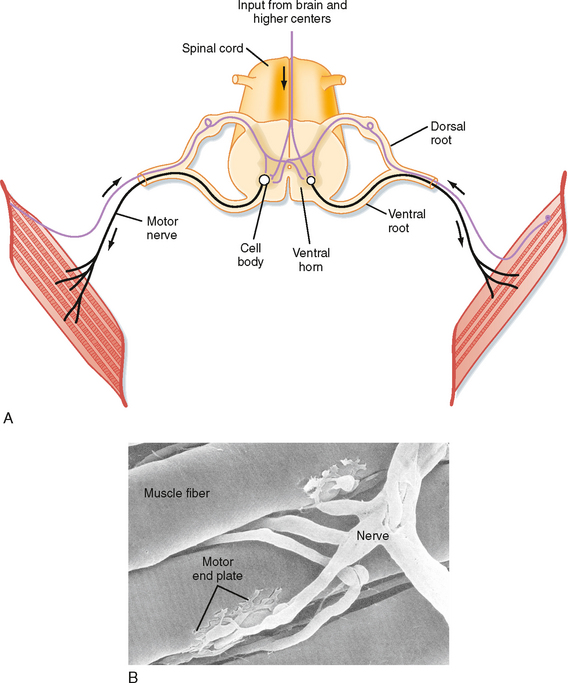
The neuromuscular junction formed by the α motor neuron is called an end plate (see Chapter 6 for details). Acetylcholine released from the α motor neuron at the neuromuscular junction initiates an action potential in the muscle fiber that rapidly spreads along its length. The duration of the action potential in skeletal muscle is less than 5 msec. This contrasts with the duration of the action potential in cardiac muscle, which is approximately 200 msec. The short duration of the skeletal muscle action potential allows very rapid contractions of the fiber and provides yet another mechanism by which the force of contraction can be increased. Increasing tension by repetitive stimulation of the muscle is called tetany (this phenomenon is described in more detail later in this chapter).
Excitation-Contraction Coupling
When an action potential is transmitted along the sarcolemma of the muscle fiber and then down the T tubules, Ca++ is released from the terminal cisternae SR into the myoplasm. This release of Ca++ from the SR raises intracellular [Ca++], which in turn promotes actin-myosin interaction and contraction. The time course for the increase in intracellular [Ca++] relative to the action potential and development of force is shown in Figure 12-8. The action potential is extremely short-lived (≈5 msec). The elevation in intracellular [Ca++] begins slightly after the action potential and peaks at approximately 20 msec. This increase in intracellular [Ca++] initiates a contraction called a twitch.
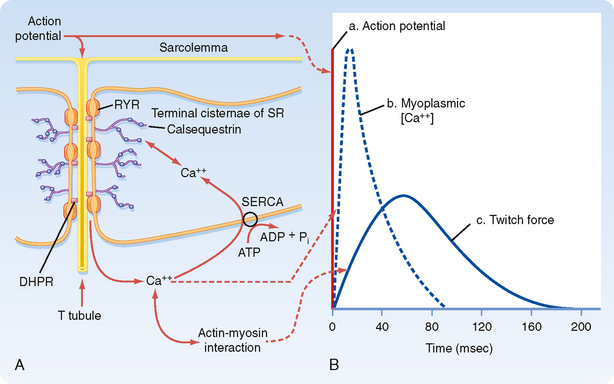
The mechanism underlying the elevation in intracellular [Ca++] involves an interaction between protein in the T tubule and the adjacent terminal cisternae of the SR. As previously described (Fig. 12-3), the T tubule represents an invagination of the sarcolemma that extends into the muscle fiber and forms a close association with two terminal cisternae of the SR. The association of a T tubule with two opposing terminal cisternae is called a triad. Although there is a gap (≈15 nm in width) between the T tubule and the terminal cisternae, proteins bridge this gap. Based on their appearance on electron micrographs, these bridging proteins are called feet (Fig. 12-9). These feet are the Ca++ release channels in the membrane of the terminal cisternae that are responsible for the elevation in intracellular [Ca++] in response to the action potential. Because this channel binds the drug ryanodine, it is commonly called the ryanodine receptor (RYR). RYR is a large protein (≈500 kDa) that exists as a homotetramer. Only a small portion of the RYR molecule is actually embedded in the SR membrane. Most of the RYR molecule appears to be in the myoplasm and spans the gap between the terminal cisternae and the T tubule (Fig. 12-10).
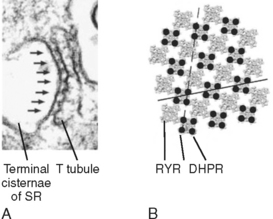
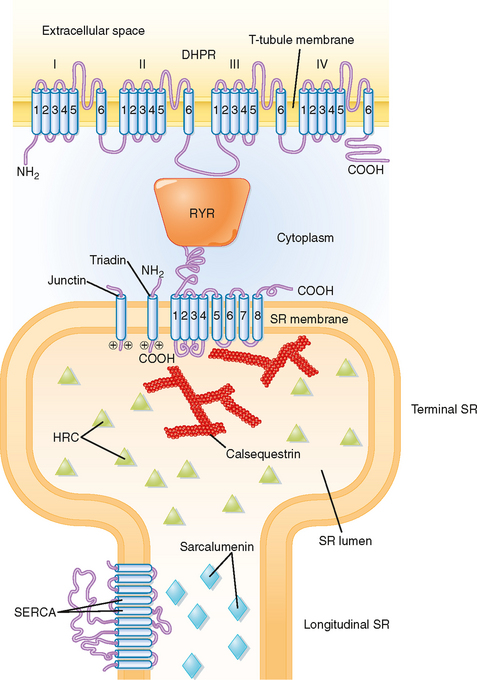
At the T-tubule membrane, the RYR is thought to interact with a protein called the dihydropyridine receptor (DHPR). DHPR is an L-type voltage-gated Ca++ channel with five subunits. One of these subunits binds the dihydropyridine class of channel blocking drugs and appears to be critical for the ability of the action potential in the T tubule to induce release of Ca++ from the SR. However, influx of Ca++ into the cell through the DHPR is not needed for the initiation of Ca++ release from the SR. Indeed, skeletal muscle is able to contract in the absence of extracellular Ca++ or with a mutated DHPR that does not conduct Ca++. Instead, release of Ca++ from the terminal cisternae of the SR is thought to result from a conformational change in the DHPR as the action potential passes down the T tubule, and this conformational change in the DHPR, by means of a protein-protein interaction, opens the RYR and releases Ca++ into the myoplasm.
Structural analysis, including the use of freeze-fracture techniques, provides evidence for a close physical association of DHPR and RYR (Fig. 12-9). DHPR in the T-tubule membrane appears to reside directly opposite the four corners of the underlying homotetrameric RYR channel in the SR membrane.
Other proteins that reside near the RYR include calsequestrin, triadin, and junctin (Fig. 12-10). Calsequestrin is a low-affinity Ca++-binding protein that is present in the lumen of the terminal cisternae. It allows Ca++ to be “stored” at high concentration and thereby establishes a favorable concentration gradient that facilitates the efflux of Ca++ from the SR into the myoplasm when the RYR opens. Triadin and junctin are in the terminal cisternae membrane and bind both RYR and calsequestrin; they could anchor calsequestrin near the RYR and thereby increase Ca++ buffering capacity at the site of Ca++ release. Histidine-rich calcium-binding protein (HRC) is another low-affinity Ca++-binding protein in the SR lumen, although it is less abundant than calsequestrin. HRC appears to bind triadin in a Ca++-dependent manner, which raises the possibility of a role greater than serving simply as a Ca++ buffer.
A variety of mutational studies have been conducted to ascertain the region of the DHPR that is critical for opening of the RYR. One possible site of interaction (depicted in Fig. 12-10) is the myoplasmic loop between transmembrane domains II and III in the α1 subunit of the DHPR. The voltage-sensing region of the DHPR involved in intramembranous charge movement is thought to reside in the S4 transmembrane segments of the α1 subunit. Genetic mutations in the RYR or DHPR, or in both, have been associated with pathological disturbances in myoplasmic [Ca++]. Such disturbances include malignant hyperthermia and central core disease, as described later. These mutations are typically observed in the myoplasmic portion of the RYR, although mutations have also been observed in a myoplasmic loop in the DHPR.
Relaxation of skeletal muscle occurs as intracellular Ca++ is resequestered by the SR. Uptake of Ca++ into the SR is due to the action of a Ca++ pump (i.e., Ca++-ATPase). This pump is not unique to skeletal muscle and is found in all cells in association with the endoplasmic reticulum. Accordingly, it is named SERCA, which stands for sarcoplasmic endoplasmic reticulum calcium ATPase. SERCA is the most abundant protein in the SR of skeletal muscle, and it is distributed throughout the longitudinal tubules and the terminal cisternae as well. It transports two molecules of Ca++ into its lumen for each molecule of ATP hydrolyzed.* Thus, the Ca++ transient seen during a twitch contraction (see Fig. 12-8) reflects release of Ca++ from the terminal cisternae via the RYR and reuptake primarily into the longitudinal portion of the SR by SERCA. The low-affinity Ca++-binding protein sarcalumenin is present throughout the longitudinal tubules of the SR and nonjunctional regions of the terminal cisternae and is thought to be involved in the transfer of Ca++ from sites of Ca++ uptake in the longitudinal tubules to sites of Ca++ release in the terminal cisternae. Recent studies suggest that sarcalumenin increases Ca++ uptake by SERCA, at least in part by buffering luminal Ca++ near the pump.
Actin-Myosin Interaction: Cross-Bridge Formation
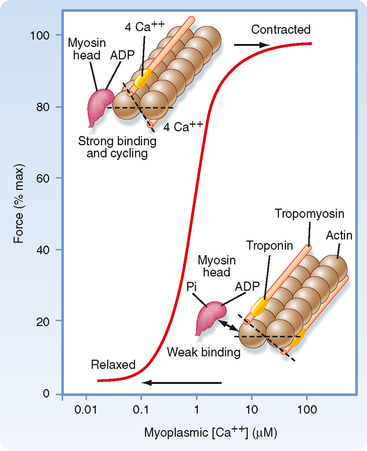
As noted, contraction of skeletal muscle requires an increase in intracellular [Ca++]. Moreover, the process of contraction is regulated by the thin filament. As shown in Figure 12-11, contractile force (i.e., tension) increases in sigmoidal fashion as intracellular [Ca++] is elevated above 0.1 μM, with half-maximal force occurring at less than 1 μM Ca++. The mechanism by which Ca++ promotes this increase in tension is as follows. Ca++ released from the SR binds to troponin C. Once bound with Ca++, troponin C facilitates movement of the associated tropomyosin molecule toward the cleft of the actin filament. This movement of tropomyosin exposes the myosin binding site on the actin filament and allows a cross-bridge to form and thereby generate tension (see later). Troponin C has four Ca++ binding sites. Two of these sites have high affinity for Ca++ but also bind Mg++ at rest. These sites seem to be involved in controlling and enhancing the interaction between the troponin I and troponin T subunits. The other two binding sites have lower affinity and bind Ca++ as its concentration rises after release from the SR. Binding of myosin to the actin filaments appears to cause a further shift in tropomyosin. Although a given tropomyosin molecule extends over seven actin molecules, it is hypothesized that the strong binding of myosin to actin results in movement of an adjacent tropomyosin molecule, perhaps exposing myosin binding sites on as many as 14 actin molecules. This ability of one tropomyosin molecule to influence the movement of another may be a consequence of the close proximity of adjacent tropomyosin molecules (Fig. 12-12).
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


