INTRODUCTION
LEARNING OBJECTIVES
After studying this chapter you should understand:
The inheritance of sickle cell disease and the difference between hemoglobin SS homozygotes, hemoglobin AS heterozygotes, and compound heterozygotes (hemoglobin SC and hemoglobin S/β-thalassemia).
The molecular basis for polymerization of deoxyhemoglobin S.
The process of sickle vaso-occlusion.
The clinical manifestations of sickle cell disease: acute pain crises and progressive organ damage.
The treatment of sickle cell disease: supportive care and prevention of sickling.
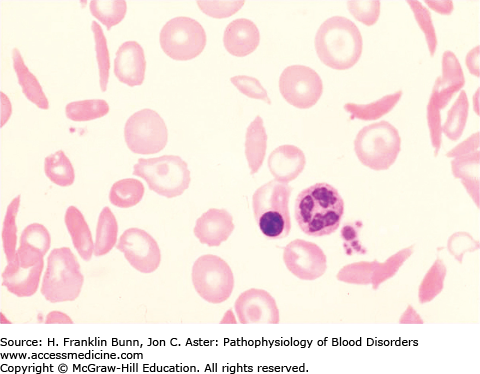
In 1910, Dr. James Herrick, while teaching a course in laboratory medicine, noted that a student from the West Indies had blood with normal-appearing red cells along with a population of “thin sickle-shaped and crescent-shaped red cells” similar to what is shown in Figure 9-1. Dr. Herrick found that this student was anemic and had breathlessness, palpitations, and occasional bouts of icterus. During the ensuing decade, a number of similar cases were reported, nearly all of them individuals of African ancestry. Most of these patients complained of intermittent attacks of severe pain. In vitro studies demonstrated that when blood from these patients was deoxygenated, all of the red cells were transformed into irregular and elongated “sickle cells.”
In the late 1940s, the era of molecular medicine was launched with the discovery by Linus Pauling that the hemoglobin from sickle cell anemia patients had an abnormal electrophoretic mobility, indicating that its structure was different from that of normal hemoglobin. Moreover, Pauling found that healthy relatives of these patients often had a 50:50 mixture of the normal and abnormal hemoglobins.
In 1957, Vernon Ingram showed that hemoglobin (Hb) S was identical to normal Hb A except for the replacement of glutamic acid, the sixth amino acid in beta (β)-globin, with valine. This was the first demonstration of a human disease arising from a single structural mutation.
GENETICS
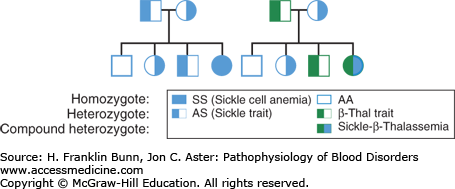
Careful observations of families showed that sickle cell anemia is inherited in an autosomal recessive manner, as shown in Figure 9-2.
FIGURE 9-2
Inheritance of sickle cell disease. A) When both parents have sickle cell trait (Hb AS) half of their offspring are expected to have sickle cell trait and one-fourth to have homozygous sickle cell anemia. B) When one parent has sickle cell trait, and the other has β-thalassemia trait, one-fourth of their offspring are expected to be compound heterozygotes (Hb S/β-thalassemia) with a symptomatic sickling disorder.
About 10% of black Americans are heterozygotes, inheriting a sickle globin gene (βS) from one parent and a normal (βA) gene from the other parent. As shown in Table 9-1, these individuals with sickle cell trait (Hb AS or AS) have no significant clinical problems unless they are subjected to severe stress, such as marked dehydration or hypoxia. Rarely AS individuals sustain splenic infarction, stroke, or sudden death following strenuous military or athletic training, particularly at high altitude. As discussed later in this chapter, the renal medulla is particularly susceptible to sickling. Many AS individuals have impaired ability to form concentrated urine (hyposthenuria), and a few have recurrent episodes of painless hematuria as a result of medullary infarction. However, damage to other organs is extremely uncommon in sickle trait individuals. They have normal life expectancy.
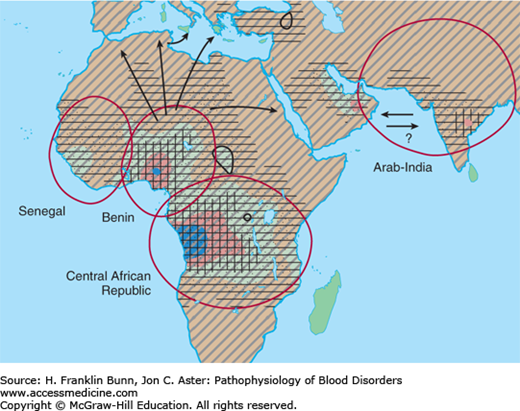
The gene frequency for Hb S is highest in Africa (Fig. 9-3), particularly in regions where malaria is endemic. Extensive analysis of polymorphisms adjacent to the β-globin gene has shown that the β6 valine mutation has arisen independently at four different times and places. This finding and the remarkably high frequency of the Hb S allele in central African populations (in which the prevalence of heterozygosity may be as high as 30%) indicate that the AS genotype confers a substantial selective advantage in this part of the world. There is strong epidemiologic evidence and somewhat less compelling experimental support for the notion that AS heterozygotes are less likely to succumb to falciparum malaria, particularly during infancy before immune defense becomes fully developed.
According to simple Mendelian genetics, about one-fourth of the children of parents with sickle trait (about 1 in 400 babies born to African American parents) will be homozygotes and have red cells containing primarily Hb S and no Hb A. As discussed in detail in this chapter, these individuals have severe hemolytic anemia and episodes of vaso-occlusion that cause acute bouts of pain and progressive organ damage. A similar clinical phenotype is encountered in individuals who are compound heterozygotes—inheriting the sickle gene from one parent and, from the other, either a β-thalassemia gene (Fig. 9-2B) or a gene that encodes Hb C, another β-globin structural mutant. Patients with S/β0 thalassemia1 are unable to make any Hb A and are as severely affected as those with Hb SS (or SS) disease. In contrast, patients with Hb S/β+-thalassemia or with Hb SC disease have less morbidity and a longer life span (Table 9-1).
MOLECULAR PATHOGENESIS
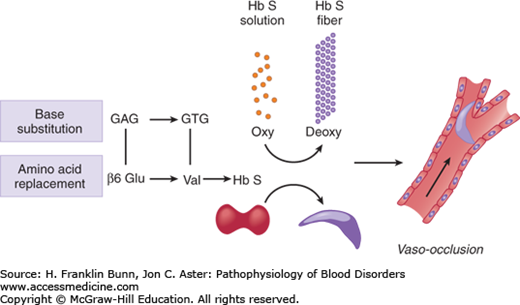
An overview of the pathogenesis of sickle cell disease is presented in Figure 9-4. The overarching question, which continues to defy easy answers, is how replacement of one of the 146 amino acids in β-globin causes a disease that can wreak havoc on nearly every organ system and display a wide range of clinical phenotypes. A partial understanding has come from rigorous study of 1) the structure of the sickle hemoglobin polymer, 2) the kinetics of polymerization, 3) the impact of recurrent episodes of sickling on the red cell membrane, and 4) the influence of Hb F on disease severity.
FIGURE 9-4
Overview of the pathogenesis of sickle cell disease. A base substitution in the β-globin gene results in the amino acid replacement β6 Glu→Val. When sickle cell hemoglobin (α2β2S) is deoxygenated, it forms a polymer that makes the red cell rigid and capable of blocking blood flow in the microcirculation.
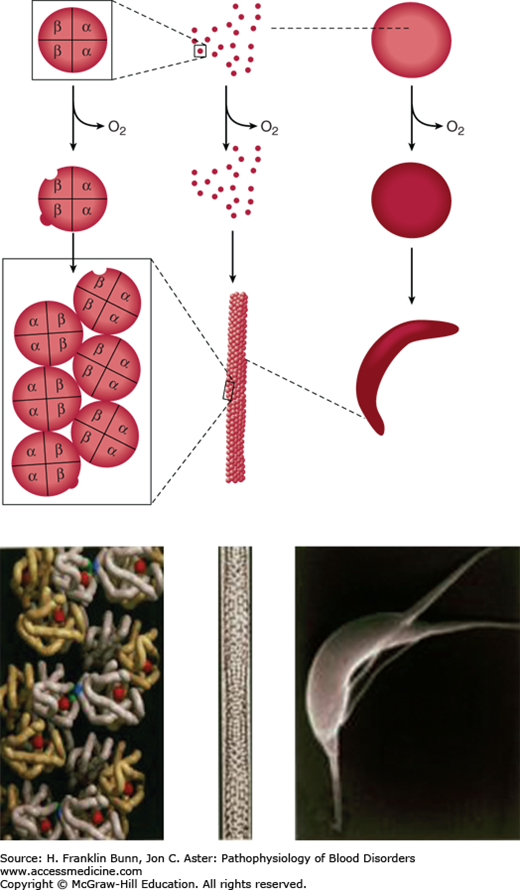
Electron micrographs of deoxygenated SS red cells reveal the presence of elongated parallel fibers of about 20 nm diameter that are oriented parallel to the long axis of sickling. Each fiber consists of a helical polymer composed of seven double strands (Fig. 9-5), each containing deoxyhemoglobin S molecules that interact through lateral and axial contacts (Fig. 9-5). Importantly, the double strand is stabilized by non-covalent hydrophobic bonding between β6 valine and a complementary “acceptor” site on another portion of the β-chain on the partner strand. In addition, the double strand is stabilized by other non-covalent contacts between molecules. When the hemoglobin is reoxygenated, conformational changes disrupt these contacts, and the polymer “melts” into individual molecules. The potential to form polymer is greatly affected by the presence of non-S hemoglobin in the red cell. Red cells of SS patients contain 2% to 20% Hb F (α2γ2), compartmentalized in a subset of cells (F cells). Because of amino acid sequence differences in gamma (γ)-globin, it cannot provide a proper hydrophobic acceptor site for binding to the β6 valine site, and therefore Hb F inhibits the polymerization of Hb S.
FIGURE 9-5
Red cell sickling and formation of the sickle polymer. Right: Deoxygenation of the red cell induces deformation into the sickle and other abnormal shapes. Left: Deoxygenation of Hb S leads to exposure of β6 Val (blue) and a complementary hydrophobic site elsewhere on the β-globin to which β6 Val binds, leading to the formation of a double strand of aggregated Hb S molecules. Middle: Seven double strands aggregate to form a 14-strand helical polymer. (Modified with permission from Bunn HF, Pathogenesis and treatment of sickle cell disease, N Engl J Med. 1997;762-769. Copyright © 1997 Massachusetts Medical Society, all rights reserved.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


