Shock
Shann D. Kim
Key Questions
• What are the common causes of cardiogenic, hypovolemic, obstructive, and distributive shock?
• What are the common cellular and tissue responses to shock of any cause?
• How does the body try to compensate for insufficient cardiac output during shock states?
• How do clinical and hemodynamic findings differ among types of shock?

http://evolve.elsevier.com/Copstead/
Shock is a life-threatening condition characterized by insufficient delivery of oxygenated blood to the microcirculation, resulting in tissue hypoxia and cellular dysfunction. In 1895, John Collins Warren described shock as a momentary pause in the act of death.1 In spite of advances in the understanding and management of shock, it still has a high rate of mortality. This chapter presents an overview of circulatory shock, including the major causes, cellular and systemic pathogenesis, clinical manifestations, and general therapeutic management.
Pathogenesis of Shock
Shock is characterized by an imbalance between oxygen supply and oxygen requirements at the cellular level.2 When the cell does not have adequate amounts of oxygen and nutrients, it is unable to meet its metabolic demands. Cellular hypoxia results in impaired cellular function and may progress to irreversible organ damage and death. The causes of circulatory shock classically are divided into four general types: cardiogenic, obstructive, hypovolemic, and distributive.3 Each of these types is associated with a number of primary causes (Box 20-1). Cardiogenic shock results from heart disorders that cause inadequate cardiac output despite sufficient vascular volume. Obstructive shock develops when circulatory blockage disrupts cardiac output, such as a large pulmonary embolus or cardiac tamponade. Because the causes of obstructive shock are associated with failure of the heart to pump sufficiently, some sources include obstructive shock within the category of cardiogenic shock. Hypovolemic shock is associated with loss of blood volume as a result of hemorrhage or excessive loss of extracellular fluids, such as through vomiting, diarrhea, or excessive diuresis. Distributive shock is characterized by a greatly expanded vascular space because of inappropriate vasodilation. Vasodilation leads to hypotension and altered perfusion of tissues. Anaphylactic, neurogenic, and septic are forms of distributive shock. Each type of shock has certain unique features (Table 20-1), but all are associated with impaired tissue oxygenation that can progress to refractory shock and organ failure.
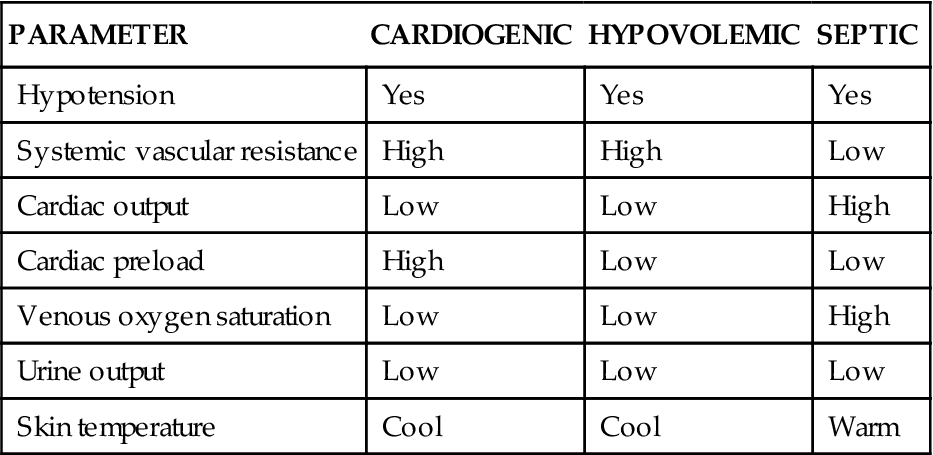
TABLE 20-1
COMPARISON OF CLINICAL FINDINGS IN DIFFERENT TYPES OF SHOCK
| PARAMETER | CARDIOGENIC | HYPOVOLEMIC | SEPTIC |
| Hypotension | Yes | Yes | Yes |
| Systemic vascular resistance | High | High | Low |
| Cardiac output | Low | Low | High |
| Cardiac preload | High | Low | Low |
| Venous oxygen saturation | Low | Low | High |
| Urine output | Low | Low | Low |
| Skin temperature | Cool | Cool | Warm |
Impaired Tissue Oxygenation
The common denominator of all forms of shock is impaired oxygen utilization by cells, which disrupts function and, if ongoing or severe, may lead to cell death, organ dysfunction, and stimulation of inflammatory reactions. Recent discoveries about the contribution of inflammatory reactions in the pathogenesis of shock have provided new insight into this complicated syndrome.
The reason for impaired oxygen utilization by cells differs with the various types of shock, but the outcomes are similar. A continuous supply of oxygen is needed by cells to allow sufficient production of energy in the form of adenosine triphosphate (ATP). Inadequate oxygen availability at the cellular level quickly impairs aerobic metabolism of glucose, fatty acids, and amino acids and causes the cells to rely on the relatively inefficient processes of glycolysis to produce cellular ATP. (A review of ATP synthesis can be found in Chapter 3.) Glycolysis is the enzymatic process of converting glucose to pyruvate, with the net production of two ATP molecules per glucose molecule. If oxygen were available, pyruvate would normally enter the mitochondria and proceed through the citric acid cycle. In the absence of cellular oxygen, the citric acid cycle is inhibited and pyruvate accumulates in the cytoplasm. Pyruvate accumulation would quickly inhibit further glycolysis and shut down ATP production entirely if not for the conversion of pyruvate to lactate. Lactate diffuses from the cell and into the extracellular fluid, and accumulation of lactate in the bloodstream (more than 5 to 6 mmol/L) is considered a sign of significant tissue hypoxia.4,5
An inadequate supply of cellular ATP inhibits energy-requiring cellular functions, including maintenance of ion concentrations across the plasma membrane. Because of their steep electrochemical gradients, extracellular sodium and calcium ions tend to leak into the cell. ATP-dependent pumps in the cell membrane are needed to continuously pump these ions back out. Failure of ion pumps leads to sodium and water accumulation in the cell (hydropic swelling) and an excess of intracellular free calcium. Intracellular calcium ions trigger a cascade of cellular events that further impair energy production and plasma membrane integrity. Cell death from oxygen deprivation takes from minutes to several hours, depending on the rate of cellular metabolic activity. However, even a short period of oxygen deprivation often sets in motion a complex cascade of events that lead to further cell damage (Figure 20-1). Two important aspects of this cascade are (1) formation of oxygen free radicals, and (2) induction of inflammatory cytokines.
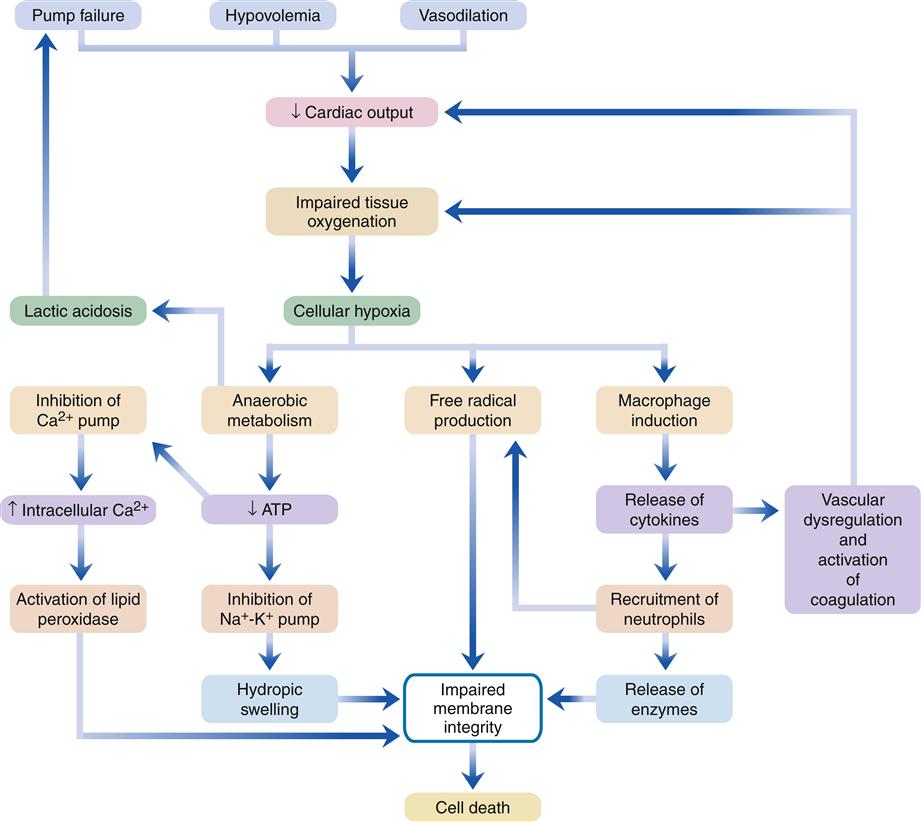
All forms of shock are associated with impaired tissue oxygenation, which triggers a cascade of events leading to tissue injury and death. ATP, Adenosine triphosphate.
Ischemic cells may produce oxygen free radicals when oxygen supplies are restored. This process has been called reperfusion injury. Reactive oxygen molecules include superoxide (O2−), peroxide (H2O2), hydroxyl radicals (OH−), and singlet oxygen (O). These molecules are unstable and will attack membrane structures, denature proteins, and damage DNA. Another source of oxygen free radicals is immune cells, particularly neutrophils, which are recruited to the area of tissue injury. Thus, cellular injury may continue and progress long after the initial hypoxic insult has been resolved.
The role that immune cytokines play in shock has been studied extensively in septic shock. The roles of these cytokines are thought to be similar in the late stages of other types of shock as well. Macrophages and tissue cells are stimulated to release inflammatory cytokines in response to hypoxic tissue injury and, in the case of septic shock, in response to endotoxin or other microorganism antigens.6 The levels of tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1) cytokines in particular have been shown to increase in the bloodstream of patients with septic shock, and these cytokines are thought to be important mediators of vascular failure and progressive organ damage.6 Numerous other immune cytokines and neurohormonal mediators have been implicated in the pathogenesis of shock (Table 20-2). These mediators represent potential therapeutic targets for a disorder that is notoriously difficult to manage effectively.
TABLE 20-2
IMMUNE CYTOKINES AND NEUROHORMONES ASSOCIATED WITH CIRCULATORY SHOCK
| MEDIATOR | ASSOCIATED DYSFUNCTION |
| IL-1α | Inflammation, vasodilation, vascular leakiness |
| IL-1β | Inflammation, vasodilation, vascular leakiness |
| IL-6 | Fever, increased acute phase protein |
| TNF-α | Inflammation, neutrophil activation |
| IL-10 | Antiinflammatory, may suppress shock |
| TGF-β | Fibrosis, pulmonary edema |
| PAF | Platelet activation, chemotaxis |
| PAI-1 | Increased clotting, thrombosis |
| Substance P | Proinflammatory |
| Chemokines | Neutrophil recruitment and binding to vessel endothelium |
| Nitric oxide | Vasodilator |
| C5a | Chemotactic |
| Protein C | Inhibits thrombus formation |
| Vasopressin | Improves vascular tone and responsiveness to NE/E |
| Cortisol | Antiinflammatory, improves vascular response to NE/E |
| Endothelin | Vasoconstriction |
| Adrenomedullin | Vasodilation |
| Norepinephrine | Vasoconstriction |
| Epinephrine | Inotropic, bronchodilation |
| Leukotrienes | Inflammation, bronchospasm |
| Histamine | Increased vascular permeability, edema |
| Heparin | Inhibits action of histamine |
| Angiotensin II | Vasoconstriction |
| Heat-shock proteins | Protect protein structure and function, inhibit apoptosis |
A hallmark of shock is failure of the microcirculation to appropriately autoregulate blood flow. Normal tissues are able to match blood flow with metabolic needs across a wide range of blood pressures. This property ensures that blood flow is evenly distributed to tissues according to metabolic needs. In shock, autoregulation fails, leading to an abnormal distribution of blood flow.7 Some capillary beds receive inadequate flow and become progressively more hypoxic, whereas other vascular routes are excessively dilated and receive too much flow. Advances in technology have allowed direct visualization of capillary flow through tissue beds and confirmed the presence of perfusion abnormalities in the microcirculation. These abnormalities differ depending on the primary cause of shock.7 In cases of reduced cardiac output resulting from hypovolemic or cardiogenic shock, there is a homogeneous reduction in blood flow through a given tissue’s arterioles, capillaries, and venules that is directly related to the severity of cardiac output reduction. In distributive shock states, such as septic shock, the degree of microcirculatory flow is heterogeneous within a tissue, with some capillaries being closed and others open. Often there is high flow rate through some of the venules and the degree of microcirculatory dysfunction is poorly correlated with systemic hemodynamics. Overall, this imbalance leads to a so-called oxygen debt in the tissues. It has been suggested that the overall oxygen debt can be estimated clinically by the serum lactate level and degree of metabolic acidosis, both of which imply a switch to anaerobic metabolism by oxygen-deprived tissues.5
In septic shock, immune cytokines are believed to be at the root of the microcirculatory maldistribution problem. TNF-α, IL-1, and other inflammatory mediators induce vascular cells to produce excessive amounts of the vasodilator nitric oxide.6 Nitric oxide in normal quantity is thought to be protective for tissues during shock, whereas excessive production is detrimental. Nitric oxide is produced in endothelial cells and vascular smooth muscle by two enzymes: nitric oxide synthase (NOS) and inducible nitric oxide synthase (iNOS). TNF-α and IL-1 increase the activity of iNOS and thereby cause excessive production of nitric oxide (Figure 20-2). Efforts to minimize the microcirculatory oxygen debt in early shock and to quickly restore adequate microcirculatory blood flow distribution, as evidenced by normalized serum lactate concentration and acid-base balance, are an important focus of therapy for all types of shock.
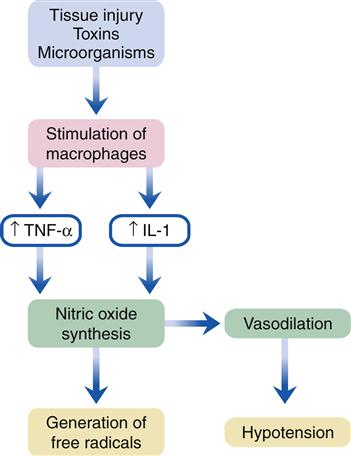
The tumor necrosis factor-α and interleukin-1 cytokines are promoters of inducible nitric oxide synthase. These cytokines are released from macrophages that have been activated by tissue injury or toxins. IL-1, Interleukin-1; TNF-α, tumor necrosis factor-α.
Compensatory Mechanisms and Stages of Shock
A number of compensatory responses are set in motion to restore tissue perfusion and oxygenation in the early stage of shock. Historically, these responses to shock have been divided into three clinical stages: compensated shock, progressive shock, and refractory shock. Although these stages may be useful for determining prognosis and the likelihood of the patient’s recovering, shock is viewed as a continuum in which compensatory mechanisms become progressively less effective as function of the microcirculation becomes increasingly impaired.
Insufficient cardiac output and decreased effective tissue perfusion are early defects in all types of shock. Insufficient cardiac output may be a consequence of an ineffective cardiac pump (cardiogenic, obstructive) or insufficient blood volume to fill the vascular space (hypovolemic, distributive). A number of compensatory mechanisms are triggered in response to inadequate cardiac output in an attempt to restore adequate perfusion pressure (Figure 20-3). Baroreceptors located in the aorta and carotid arteries quickly sense the decrease in pressure and transmit signals to the vasomotor center in the brainstem medulla. Stimulation of the sympathetic nervous system (SNS) results in increased cardiac output and vascular resistance. Because blood pressure is determined by the product of cardiac output and vascular resistance, an increase in one or both of these factors will help restore blood pressure. The SNS increases cardiac output through several mechanisms. The adrenal medulla is stimulated to release increased amounts of the catecholamines epinephrine and norepinephrine (NE), which circulate to the heart and stimulate β1 receptors. The β1 receptors respond by increasing the heart rate and force of contraction in an attempt to increase cardiac output. The SNS also enhances venous return to the heart by constricting systemic arterioles and venules. Arterial vasoconstriction reduces flow through the capillary bed, which causes hydrostatic pressure in the capillaries to fall. Fluid reabsorption from interstitial spaces helps increase blood volume and improve preload. Blood vessels in the skin, kidneys, and gastrointestinal tract constrict and shunt blood to the heart and brain.
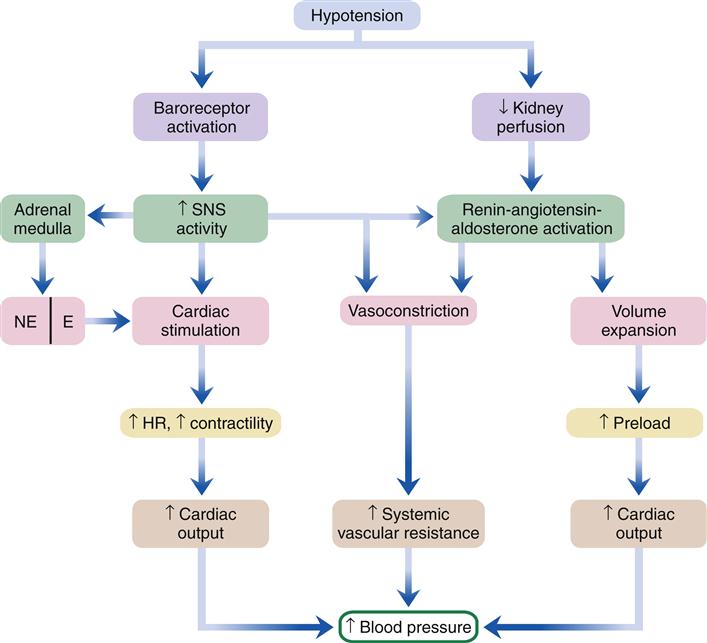
E, Epinephrine; HR, heart rate; NE, norepinephrine; SNS, sympathetic nervous system.
The SNS stimulates cells in the kidney to release renin, which triggers the renin-angiotensin-aldosterone system (RAAS). Renin is also secreted from the kidneys in response to decreased blood flow and pressure in the afferent arterioles. Renin triggers the formation of angiotensin II, which is a potent vasoconstrictor and also stimulates kidney nephrons to conserve sodium and water. Conservation of volume by the kidney is further enhanced by aldosterone, which is secreted from the adrenal cortex in response to angiotensin II (Ang II). Reabsorption of fluid from the kidney helps increase blood volume and enhances venous return to the heart. Another hormone, antidiuretic hormone (vasopressin), is secreted from the posterior pituitary in response to reduced blood volume. Antidiuretic hormone stimulates the kidney tubules to reabsorb water and improves the vascular response to catecholamines. In shock, urine output may fall to zero as the kidneys attempt to conserve fluid to maintain blood volume and cardiac output. Unfortunately, the kidney tubules often sustain damage because of the low-flow state, which may result in the complication of acute renal failure.
These compensatory mechanisms work well in the early stage of hypovolemic shock and may maintain blood pressure within the normal range until the volume of blood loss becomes too great (Figure 20-4). In other forms of shock, compensatory mechanisms are less effective in restoring cardiac output. In cardiogenic shock, the compensatory responses may worsen the already high preload and impose a greater workload on the failing heart. In distributive shock, the vasculature is not responsive to SNS signals to constrict. Blood pools in the peripheral tissues, which makes it difficult for the heart to maintain cardiac output despite sympathetic stimulation to increase the heart rate and contractility.
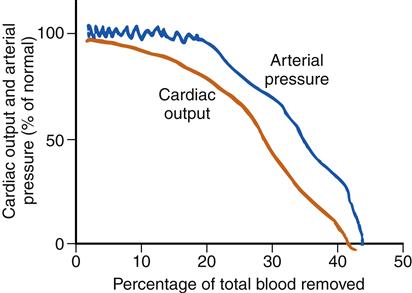
When volume losses equal about 25% of the total blood volume, blood pressure falls precipitously. (Redrawn from Hall JE: Guyton and Hall textbook of medical physiology, ed 12, Philadelphia, 2011, Saunders, p 274.)
The early, compensated stage of shock may be difficult to detect clinically (Figure 20-5). A high index of suspicion is needed in patients with heart failure, trauma, blood loss, and severe infection. In addition, the following clinical findings may be present:
• A narrow pulse pressure with or without hypotension
• Tachycardia greater than 100 beats/min
• Increased urine specific gravity
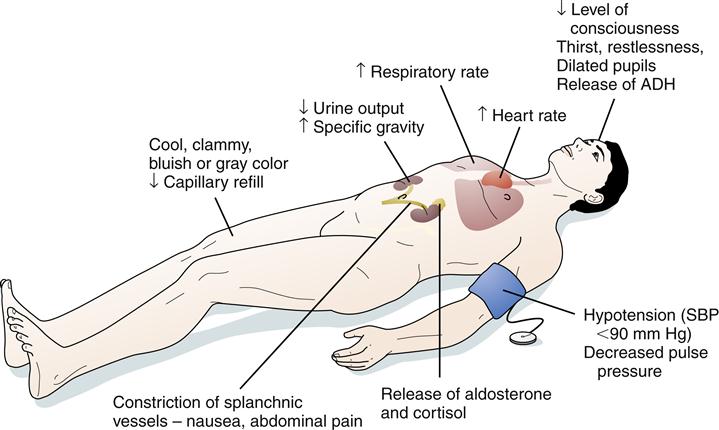
ADH, Antidiuretic hormone; SBP, systolic blood pressure.
At some point, which is highly variable and differs among individuals depending on age, comorbidities, and specific etiology, the compensatory mechanisms can no longer sustain adequate perfusion to tissues and cells begin to suffer significant hypoxic injury. This condition is sometimes called the progressive stage of shock. Active therapeutic intervention is required at this stage or the patient will probably not survive. As previously described, reduced delivery of oxygen to tissues results in hypoxic injury, free radical damage, and stimulation of the inflammatory response. Lactic acidosis may occur during the progressive stage of shock. In addition to being a marker of anaerobic metabolism, lactate can alter the acid-base balance of the blood and create metabolic acidosis. Metabolic acidosis places a greater burden on the respiratory and renal systems and may contribute to further dysfunction. Metabolic acidosis can affect electrolyte balance and contribute to cardiac dysrhythmias and conduction disturbances. In addition, myocardial depressant factors are released that impair myocardial contractility.8 These factors contribute to reduced cardiac output and a progressive cycle of worsening tissue hypoxia.
As shock continues to progress, the vascular system begins to fail. Arterioles become unresponsive to catecholamines and previously constricted vascular beds begin to dilate. Widespread dilation and low cardiac output combine to produce severe hypotension. The low blood pressure is not sufficient for organ perfusion. At this stage the effects of shock produce more shock processes. Tissue damage often activates the clotting cascade, which contributes to sluggish blood flow, vascular thrombosis, and more severe tissue ischemia. Release of inflammatory mediators, along with vascular occlusion, may precipitate organ failure. The kidney, liver, and lung are particularly susceptible. At some point, the stage of refractory shock occurs and the patient becomes unresponsive to therapeutic interventions.
The progressive stage of shock is characterized by the following clinical manifestations:
• Low blood pressure, usually lower than 90 mm Hg
• Acute renal failure (e.g., oliguria, increased levels of blood urea nitrogen and serum creatinine)
• Decreased level of consciousness
Types of Shock
In addition to the general pathophysiology of shock just described, each type of shock has special features that affect prevention, diagnosis, and treatment. These features are briefly reviewed here, and the reader is referred to the specific chapters that describe the primary disorders that predispose to shock, including cardiogenic (Chapters 18 and 19), hypovolemic (Chapter 24), anaphylactic (Chapter 10), neurogenic (Chapter 44), and septic (Chapter 8).
Cardiogenic Shock
Etiology and pathogenesis
Cardiogenic shock occurs primarily as a result of severe dysfunction of the left, right, or both ventricles that results in inadequate cardiac pumping. The most common cause of cardiogenic shock is myocardial infarction resulting in a significant dysfunction or loss (greater than 40%) of left ventricular myocardium.9 Other causes of cardiogenic shock include right ventricular myocardial infarction, end-stage cardiomyopathy, papillary muscle dysfunction, free wall rupture, and congenital heart defects.
The low cardiac output state is associated with a high left ventricular diastolic filling pressure (preload), a finding that differentiates cardiogenic from hypovolemic forms of shock (Figure 20-6). High left ventricular preload leads to movement of fluid from the pulmonary vascular beds into the pulmonary interstitial space, which initially results in interstitial pulmonary edema and later in alveolar pulmonary edema.
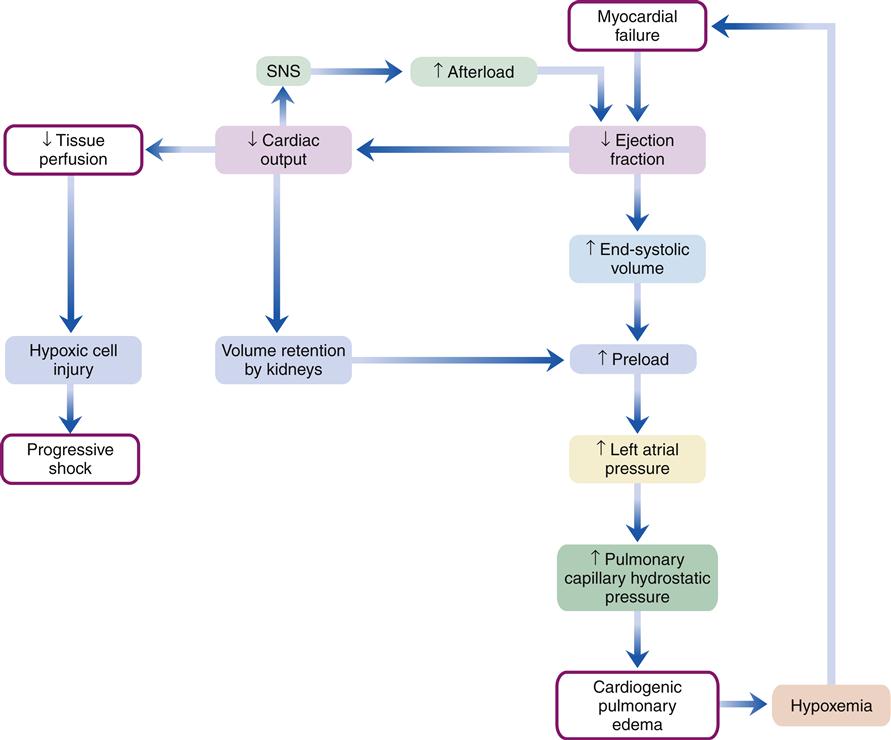
SNS, Sympathetic nervous system.
The SNS is stimulated as a compensatory mechanism to increase cardiac output. The result is an increase in heart rate and systemic vascular resistance. High systemic vascular resistance increases the workload on the heart. Activation of the RAAS results in further increases in resistance and preload. The net effect of the activation of compensatory mechanisms is to increase myocardial workload and oxygen demand. Consequently, the compensatory responses can precipitate further cardiac damage and cause a progressive decline in cardiac output.
Clinical manifestations
Sympathetic nervous stimulation increases the heart rate and vascular resistance, which maintain blood pressure even though cardiac output has decreased. As compensatory mechanisms fail, systolic blood pressure falls and diastolic pressure increases (as a result of the sympathetic stimulation), thus narrowing the pulse pressure. Heart rates exceed 100 beats/min. Peripheral vasoconstriction occurs and produces cool, clammy skin. Auscultation of the lungs reveals coarse crackles resulting from pulmonary edema. An S3 summation gallop may be audible over the left apex as a result of increased preload in the left ventricle.
Frequent assessments of cardiac output and cardiac index are helpful in the clinical treatment of a patient in cardiogenic shock. A pulmonary artery catheter may be inserted to measure cardiac index and left heart filling pressures (e.g., pulmonary capillary occlusion pressure). Pulmonary artery pressures are increased, with the pulmonary capillary occlusion pressure typically being greater than 15 mm Hg (normal, less than 12 mm Hg).9 When pulmonary capillary occlusion pressure acutely increases, pulmonary congestion may develop because fluid shifts from the capillary into the interstitial and alveolar spaces. Patients with chronic congestive heart failure may not develop pulmonary edema until pulmonary occlusion pressures exceed 20 to 40 mm Hg. Arterial blood gas values initially demonstrate a respiratory alkalosis secondary to hyperventilation. As pulmonary edema progresses, respiratory acidosis with hypoxemia may occur. Hypoxemia further impairs myocardial function.
Determination of mixed venous oxygen levels in blood samples obtained from the pulmonary artery catheter is helpful in assessing the adequacy of cardiac output. Decreased tissue oxygen delivery because of low cardiac output increases the degree of oxygen extraction. Mixed venous blood samples show decreased venous oxygen saturation (SvO2) with a decreased cardiac output. An increase in mixed venous oxygenation would be expected with improved cardiac output.
Treatment
Cardiogenic shock is difficult to manage because the underlying myocardial damage is often not reversible. Prevention of cardiogenic shock through measures to limit infarct size during acute myocardial ischemia is desirable. Early efforts to restore coronary perfusion are associated with a decrease in the incidence of cardiogenic shock after myocardial infarction.9,10 (A discussion of reperfusion therapy can be found in Chapter 18.)
The goal of treatment for cardiogenic shock is to decrease myocardial oxygen demands, increase myocardial oxygen delivery, and increase cardiac output. It is difficult to achieve these goals because interventions to increase cardiac output tend to increase myocardial oxygen demands.
Pharmacotherapy
Positive inotropic drugs are frequently used in the management of cardiogenic shock to increase contractility. Positive inotropes include β-adrenergic agonists such as NE, dobutamine, and dopamine and phosphodiesterase inhibitors that prevent the degradation of cyclic adenosine monophosphate (cAMP). These drugs have the ability to increase contractility, increase cardiac output, and increase tissue perfusion; however, these drugs increase myocardial oxygen demand. NE is the natural neurotransmitter of the sympathetic nerves and mimics SNS activation by increasing heart rate, contractility, and vascular resistance. Dobutamine increases contractility by stimulating β receptors. However, unlike NE or dopamine, dobutamine has minimal α-receptor activity. The major effect of dobutamine is on contractility rather than heart rate. Dobutamine may contribute to a decrease in vascular resistance and must be used with caution in hypotensive patients.11 Vasodilators may be used to decrease the workload of the heart by decreasing left ventricular afterload. Examples of commonly used vasodilators include nitroprusside and nitroglycerin.
Mechanical assist devices
Cardiogenic shock is sometimes managed by mechanical assist devices. For temporary management, intraaortic balloon counterpulsation may be indicated.12 A catheter with a balloon at the distal segment is inserted through the femoral artery and positioned in the aorta just distal to the left subclavian artery. The balloon is connected to a console that triggers the balloon to inflate in diastole and deflate in systole. The effect of balloon inflation during diastole is to increase perfusion pressure of the coronary arteries. Sudden deflation of the balloon just before ventricular systole creates a vacuum effect in the aorta that reduces left ventricular afterload. A reduction in afterload decreases left ventricular workload and increases stroke volume. Balloon counterpulsation restricts mobility and is associated with a number of vascular complications. Long-term management of patients with low cardiac output can be achieved with mechanical pumps that take over the function of the ventricle or ventricles (ventricular assist devices). Ventricular assist devices (VADs) are commonly used in patients waiting for a heart transplant. In some cases, the temporary decrease in cardiac workload afforded by the VAD is associated with significant improvement in cardiac structure and function, and the device can be removed.



