CHAPTER 26 Second Messengers
This chapter considers the remarkable variety of small molecules that carry signals inside living cells. These second messengers are chemically diverse, ranging from hydrophobic lipids confined to membrane bilayers, to an inorganic ion (Ca2+), to nucleotides (cyclic adenosine monophosphate [cAMP] and cyclic guanosine monophosphate [cGMP]), to a gas (nitric oxide). The messages that these molecules carry are encoded by their concentrations. In the simplest case, a rise or fall in the concentration of the second messenger conveys a signal from its source to its target. In other cases, the signal depends on the rate or frequency of the fluctuations in the concentration of the second messenger. The local concentration of a second messenger depends on the rate of production, the rate of diffusion from the site of production, and the rate of removal. Most second messengers are produced by enzymes that can switch on and off rapidly, allowing modulation of the concentration of second messengers on a millisecond time scale. In the case of Ca2+, the cytoplasmic concentration is determined by channels that release the ion from membrane-delimited stores and by pumps that remove it from cytoplasm.
The complexity of the signaling pathways is determined by the number of sources and the number of targets of each second messenger. Generally, multiple signal sources and multiple second messenger targets generate more complexity than one can fully appreciate. This chapter deals with this complexity only in passing. Chapter 27 considers a few model systems in which it is possible to understand how signals are integrated and transduced.
Cyclic Nucleotides
Two cyclic nucleoside monophosphates—adenosine 3′,5′-cyclic monophosphate (cAMP) and guanosine 3′,5′-cyclic monophosphate (cGMP)—are employed as second messengers (Fig. 26-1). Both act by binding reversibly to specific proteins. Enzymes that produce and degrade cyclic nucleotides determine the concentrations of these messengers available to bind targets. These enzymes turn over substrates rapidly, so they can amplify signals massively on a millisecond time scale, under the control of diverse signaling pathways (see Chapter 27). Cyclases make cyclic nucleotides in a single step from the corresponding nucleoside triphosphate, either adenosine triphosphate (ATP) or guano-sine triphosphate (GTP).
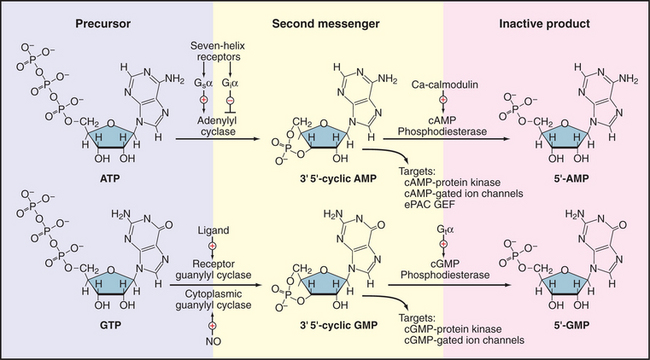
Figure 26-1 cyclic nucleotide metabolism. Synthesis and degradation of cAMP and cGMP, including regulatory inputs and targets. Gsα, Giα, and Gtα are trimeric GTPase a subunits (see Fig. 25-9). C A, calcium; NO, nitric oxide.
Cyclic nucleotides diffuse in the cytoplasm at about the same rate as in free solution, activating a modest repertoire of downstream targets, including protein kinases (see Figs. 25-3 and 25-4), cyclic nucleotide–gated ion channels (see Fig. 10-10), and, in the case of cAMP, one class of nucleotide exchange factors (Epac) for small GTPases (Rap1 and Rap2). The components of this system are quite ancient, since the protein domains that bind cAMP or cGMP are homologous to the cAMP-binding domain of CAP, a bacterial transcription factor. Enzymes called cyclic nucleotide phosphodiesterases degrade cAMP and cGMP to inactive nucleoside 5′-monophosphates. Eleven genes encode more than 40 different phosphodiesterases, which vary in their specificities for the two cyclic nucleotides, expression in various tissues, and localization to cellular compartments.
A family (10 human genes) of enzymes called adenylyl cyclases synthesize cAMP from ATP. Multiple transmembrane segments anchor most of these enzymes to the plasma membrane. Two homologous catalytic domains reside in the cytoplasm (C1 and C2 in Fig. 26-2). These cytoplasmic domains can be produced experimentally as soluble proteins separately from the transmembrane domains and can then be recombined to make a fully active enzyme. Both domains are necessary because the active site lies at the interface of the two domains. One adenylyl cyclase is a soluble enzyme that can concentrate in the nucleus. Generally, the concentration of adenylyl cyclases is very low relative to the trimeric G-proteins that regulate their activity.
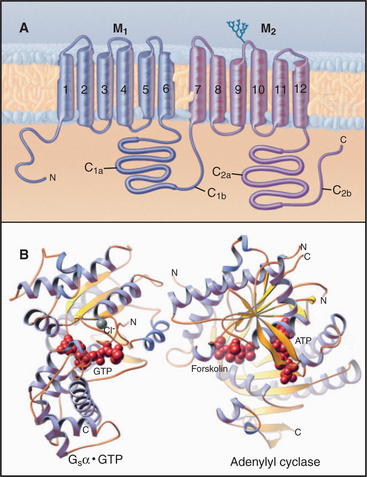
Multiple regulatory mechanisms act synergistically to regulate adenylyl cyclases. GTP-Gsα the GTPase subunit of a trimeric G-protein, activates many membrane-associated adenylyl cyclases by binding far from the active site (Fig. 26-2) and inducing a conformational change. Ca2+-calmodulin or protein kinase C (PKC) activates some adenylyl cyclases. GTP-Giα the GTPase subunit of another trimeric G-protein, or protein kinase A (PKA) inhibits some cyclases. Gβγ subunits of trimeric G-proteins activate some adenylyl cyclases but inhibit others. These diverse regulatory mechanisms allow adenylyl cyclases to integrate a variety of input signals. The diterpene forskolin from a Coleus plant binds to and activates adenylyl cyclases. Forskolin is useful experimentally for manipulating the cAMP concentration in cells and, on the basis of its ability to bind adenylyl cyclases, was essential in the initial purification of the enzyme by affinity chromatography. Regulation of a soluble form of adenylyl cyclase differs from that of all other isoforms. It is activated by bicarbonate, an essential step in sperm maturation.
In resting cells, the concentration of cAMP is so low, approximately 10−8M, that it does not bind its targets. Stimulation of appropriate receptors (such as the seven-helix β-adrenergic receptor, see Fig. 27-3) increases the cytoplasmic cAMP concentration more than 100-fold, enough to saturate the PKA regulatory subunits (Fig. 26-3). Dissociation of the regulatory (R) subunits frees the active PKA catalytic subunits to phosphorylate cytoplasmic and membrane substrates and to move into the nucleus to activate the transcription factor CREB (cyclic nucleotide regulatory element–binding protein) (see Fig. 15-22).
Outside the animal kingdom, cAMP has many functions. In bacteria, cAMP controls gene expression in response to nutritional conditions. The cellular slime mold Dictyostelium uses cAMP as an extracellular signal, acting through a seven-helix receptor, for its social interactions (see Fig. 38-12).
Guanylyl cyclases are dimeric enzymes similar to adenylyl cyclases. In fact, mutation of just two amino acid residues can convert a guanylyl cyclase to an adenylyl cyclase. Vertebrates express two types of guanylyl cyclases (see Fig. 24-9). A family of transmembrane receptors with cytoplasmic cyclase domains respond to ligand binding by producing cGMP. Cytoplasmic guanylyl cyclases are activated by nitric oxide and carbon monoxide, which bind a heme group in a regulatory domain (see the section titled “Nitric Oxide” later in this chapter).
Lipid-Derived Second Messengers
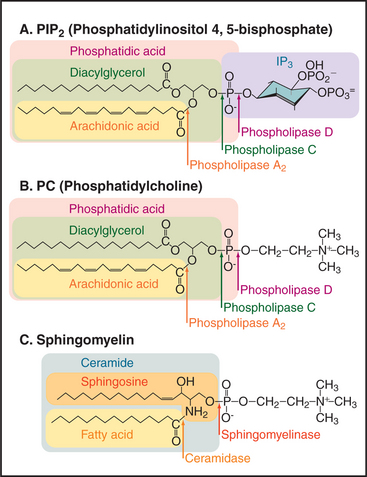
Phosphoglycerides (see Fig. 7-2) and sphingolipids (see Fig. 7-3) not only form cellular membranes but also participate in a wide range of signaling mechanisms. The long list of intracellular and extracellular second messengers derived from lipids will undoubtedly expand in the future. Three membrane lipids are the primary sources of these signaling molecules (Fig. 26-4):
Enzyme Reactions That Produce Lipid Second Messengers
Three types of enzyme reactions generate lipid second messengers:
Cells also use transferases that add or exchange head groups or fatty acids on membrane phosphoglycerides (see Fig. 7-2). These enzymes are essential for lipid biosynthesis but have not been implicated in signaling. For example, transferases add choline (as the nucleotide conjugate cytidine diphosphate [CDP]–choline) to DAG to make phosphatidylcholine). In the case of phosphatidylinositol, the head group is provided by dephosphorylation of IP3 to inositol, which is recombined enzymatically with a CDP conjugate of DAG to make phosphatidylinositol.
Sequential action of two or more enzymes produces some lipid second messengers. For example, cells can make DAG in two steps from phosphatidylcholine: PLD first makes phosphatidic acid, which is dephosphorylated by phosphatidic acid phosphatase. PLD followed by PLA2 makes lysophosphatidic acid from phosphatidylcholine. PLA2 also initiates the production of a huge family of fatty acid derivatives called eicosanoids by cleaving the fatty acid arachidonic acid from phosphatidylcholine. Cyclooxygenases and lipoxygenases then modify arachidonic acid to make prostaglandins, thromboxanes, and leukotrienes (Figs. 26-9 and 26-10), which leave the cell to interact with cell surface receptors.
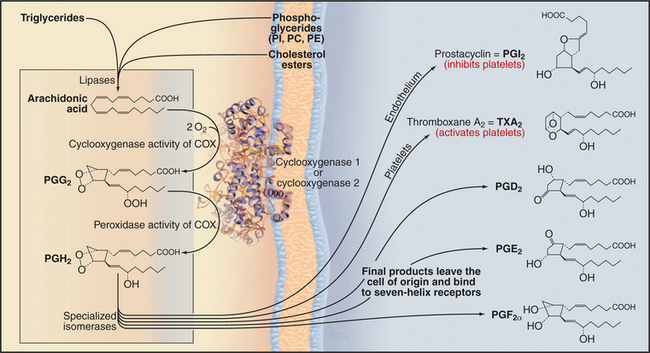
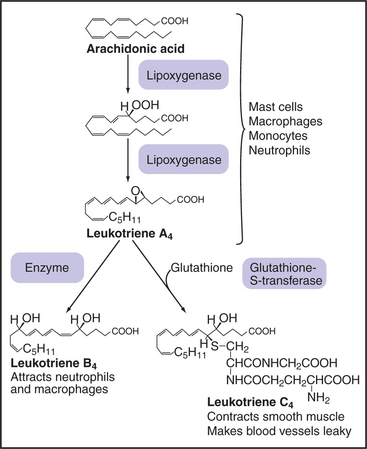
Targets of Lipid Second Messengers
Downstream targets of the lipid second messengers are diverse but can be generalized to some extent into three categories (Fig. 26-5):
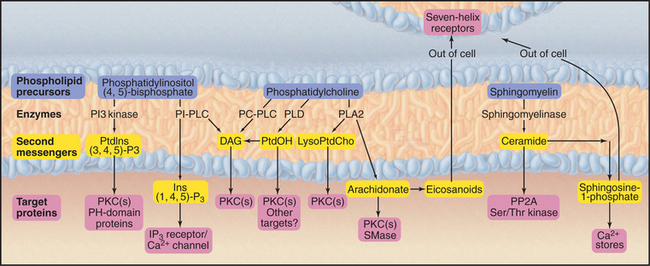
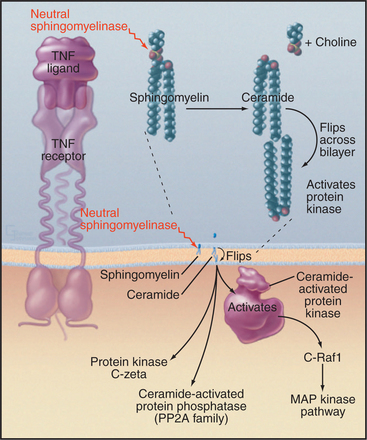
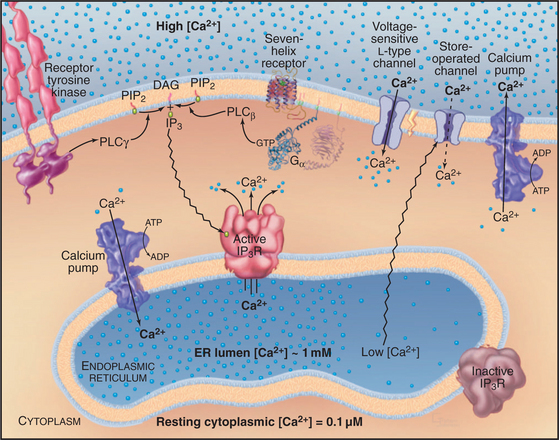
Lipid second messengers participate in many processes that are considered elsewhere in the book. For example, regulated secretion (see Chapter 21) in response to an agonist binding a seven-helix or tyrosine kinase receptor requires a transient Ca2+ signal in the cytoplasm. The Ca2+ is released from the membrane stores by IP3, which is produced by the action of a PI-PLC on PIP2. IP3-mediated Ca2+ release from the endoplasmic reticulum also controls smooth muscle contraction (see Fig. 39-21).
Protein Kinase C
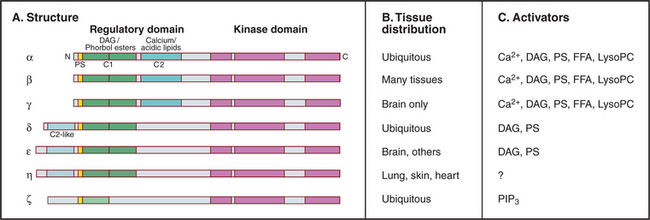
Many lipid second messengers, including DAG, PIP3, arachidonic acid, phosphatidic acid, and lysophosphatidylcholine activate one or more of the 10 protein kinase C (PKC) isozymes expressed by vertebrate cells (Fig. 26-6). These multiple PKC isozymes provide a selective response to various lipid second messengers. Some, but not all, PKC isozymes also require Ca2+ for activation. Sphingosine may inhibit some PKC isozymes.
PKCs have a protein kinase catalytic domain (see Fig. 25-3) and N-terminal regulatory domains. A pseudosubstrate sequence binds intramolecularly to the active site and inhibits activity by blocking substrate binding. Pseudosubstrates have alanine at the phosphorylation site instead of serine as substrates have. During apoptosis, caspases cleave off this regulatory domain (see Fig. 46-10) producing constitutively active PKC isoforms.
PKCs can provide either positive or negative feedback to the signaling pathways that turn them on. PKC activates PLD and PLA2 and provides positive feedback, because those enzymes produce more DAG to sustain the activation of PKC. On the other hand, PKC provides negative feedback when it phosphorylates and inhibits both growth factor receptors and PI-PLCγ1. PKC also phosphorylates and inhibits PI-PLCβ, generating negative feedback after activation of seven-helix receptors. Negative feedback makes both of these signaling events transient.
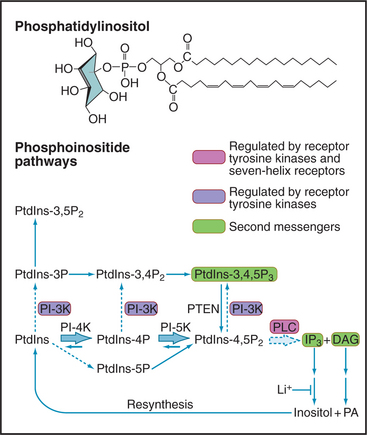
Phosphoinositide Signaling Pathways
Although minor in terms of mass in biological membranes, phosphoinositides are major players in signal-ing (Fig. 26-7). The parent compound, phosphatidyl-inositol (PI), is a phosphoglyceride with a cyclohexanol head group called inositol. Specific lipid kinases phosphorylate the 4 and 5 hydroxyl groups of phosphatidylinositol to form PI(4-)P and PI(4,5-)P2, usually referred to simply as PIP and PIP2. A specific phosphatase can remove the D5 phosphate. Inactivation of the single copy of this gene on the X-chromosome causes Lowe’s syndrome with cataracts, renal failure, and mental retardation.
PIP2 is a substrate for a family of receptor-controlled PI-PLCs that cleave off the phosphorylated head group, producing two potent second messengers: IP3 and DAG. Water-soluble IP3 activates Ca2+ release channels in the endoplasmic reticulum (Fig. 26-12), and lipid-soluble DAG activates PKC. In contrast to Ca2+, which diffuses slowly and acts locally, IP3 diffuses rapidly through the cytoplasm, triggering Ca2+ release. DAG is confined to membranes but diffuses laterally to bind and activate PKC. Enzymes inactivate both IP3 and DAG. DAG is inactivated by phosphorylation to make phosphatidic acid, a second messenger in its own right but also an intermediate in the resynthesis of phosphoinositides. IP3 is dephosphorylated to inositol, which is inactive as a second messenger. LiCl inhibits the final step: dephosphorylation of inositol-1-phosphate. Remarkably, Li+ is clinically useful as a treatment for bipolar disorder, presumably by interfering with phosphoinositide signaling in the brain. In some tissues, IP3 is inactivated by phosphorylation to inositol 1,3,4,5-tetrakisphosphate (IP4).
Vertebrates use 10 classes of PI-PLCs to provide tissue-specific coupling of various receptors to the production of IP3 and DAG. PI-PLCγ1, expressed in the brain, is activated by tyrosine phosphorylation when its SH2 domains bind a tyrosine-phosphorylated receptor tyrosine kinase (Fig. 26-12). The trimeric G-protein Gqα activates another isozyme, PI-PLCβ.
Receptor tyrosine kinases activate another family of lipid kinases, PI-3 kinases, which phosphorylate the 3-hydroxyl group of PI, PIP, and PIP2. The products are PI(3-)P, PI(3,4-)P2, and PI(3,4,5-)P3. PI(3,4,5-)P3 is not a substrate for the PI-PLCs that produce IP3 and DAG, but it activates some PKC isozymes and binds specifically to certain pleckstrin homology domains (see Fig. 25-11), bringing enzymes such as protein kinase B (PKB/Akt) to the plasma membrane. Association with the membrane leads to phosphorylation and activation of PKB as part of insulin signaling (see Fig. 27-7). A phosphatase that removes the D3 phosphate from PIP3 is a tumor suppressor called PTEN (phosphatase and tensin homolog deleted on chromosome 10). Loss of PTEN function in tumors allows PIP3 to build up, activating PKB and growth promoting downstream pathways in many tumors. A steroid-like molecule called wortmannin inhibits PI-3 kinase relatively specifically and is used to investigate the physiological roles of PIP3.



