Sarcoma
FREDERICK C. KOERNER
Mammary sarcomas are a heterogeneous group of malignant neoplasms that arise from the mammary stroma. Excluded from this chapter are malignant lymphomas and malignant phyllodes tumor (MPT), which are covered in other chapters. Lesions at the most low-grade end of the spectrum of fìbroblastic tumors constitute the group of neoplasms discussed in Chapter 38 under the heading of “fibromatosis.” Although mammary fibromatosis does not metastasize, it can pursue an inexorable course of local recurrence, resulting in extensive local destruction and even death as a consequence of visceral invasion. Neoplasms included in the category of mammary sarcoma, or stromal “sarcoma,” are thought to arise from interlobular mesenchymal elements, which constitute the supporting mammary stroma.
A diagnosis of mammary sarcoma can be established only after metaplastic carcinoma is excluded. The distinction is important for treatment as well as for prognosis. The lesion should be sampled extensively for evidence of in situ or invasive carcinoma. Perhaps the most difficult distinction in this regard lies between fibrosarcoma and a metaplastic spindle cell carcinoma, which has no discernible epithelial component. Immunohistochemical studies for epithelial and myoepithelial markers are useful for detecting evidence of epithelial origin or inconspicuous foci of epithelial cells in a carcinoma that has undergone a virtually complete conversion to a spindle cell neoplasm (see Chapter 16).
The diagnosis of mammary sarcomas should be reported in the same histogenetic terms used for soft part sarcomas, which occur throughout the body. The relative frequency of specific types of breast sarcomas is difficult to determine from the literature because these lesions have sometimes been referred to by the general term “stromal sarcoma.” However, some differences in frequency do appear to exist. For example, one of the most common forms of mammary sarcoma, angiosarcoma, is proportionately less common among somatic sarcomas. Other distinct mammary sarcomas include leiomyosarcoma, liposarcoma, sarcomas with bone and cartilage, malignant fibrous histiocytoma (MFH) and fibrosarcoma, rhabdomyosarcoma, and other rare types.
Mammary sarcomas arise only rarely. According to data from the Surveillance Epidemiology and End Results (SEER) database, the annual incidence is 4.6 cases per million women.1 Several factors predispose patients to the development of mammary sarcomas. Women treated for breast carcinoma experience a slight increase in their risk of softtissue sarcomas,2,3 and the use of irradiation further increases the risk of the development of angiosarcomas, pleomorphic undifferentiated carcinomas, and other rare types of sarcoma within the field of irradiation.2,3,4 The risk of developing postirradiation sarcoma increases significantly 3 years after the diagnosis of carcinoma, peaks at approximately 10 years, and declines to approximately the risk seen in patients who did not receive irradiation after 23 years.4 Rare mammary sarcomas have developed in association with cosmetic implants or foreign material,5 but the evidence does not prove a causal relationship between the presence of these substances and the development of the sarcoma.
Women make up the vast majority of patients with mammary sarcomas, and they accounted for 98.5% of the cases of sarcoma in the 19 studies tabulated by Al-Benna et al.,6 some of which included cases of MPTs and metaplastic carcinomas. These data may not account for all cases affecting men, because certain tumors in men may have been classified as sarcomas of the chest wall rather than of the breast. Females of all ages are affected. In the largest series,7 the patients’ ages range from 13 to 86 years, and the median age is 55 years. Using information from published studies, Al-Benna et al.6 calculated a weighted mean age of 50.0 years.
As a group, mammary sarcomas vary greatly in size, ranging from less than 1 to 30 cm or more. In most studies, the mean and median sizes fall between 4 and 7 cm. The authors of one large series7 reported a median tumor size of 4.45 cm. The gross appearance of the tumors is influenced in part by the specific histologic characteristics of the lesion, but the specimens typically consist of fleshy, moderately firm, pale tissue with varying amounts of hemorrhage and necrosis (Fig. 39.1). Most sarcomas appear well circumscribed grossly, even if the border is invasive histologically.
The prognosis for patients with mammary sarcomas varies somewhat depending on certain characteristics of the sarcoma. For example, high-grade angiosarcomas have an especially unfavorable prognosis, whereas the outcomes of patients with other histologic types of sarcoma do not differ significantly. The size of the sarcoma may also provide prognostic information, but the data regarding this relationship vary. In a group of 83 women with primary breast sarcomas studied by Zelek et al.,1 with a median follow-up of 7.8 years, the 10-year overall survival (OS) and disease-free survival
(DFS) rates were 62% and 50%, respectively. Tumor size (less than 5 cm, 5 to 10 cm, or larger than 10 cm) was significantly related to 10-year DFS. Another series of 25 women with primary breast sarcomas and a mean follow-up of 10.5 years was reported by Adem et al.8 Local recurrence occurred in 11 patients after a mean follow-up of 15 months, and 10 patients (40%) had systemic metastases. The 5-year DFS was 90% for tumors 5 cm or less and 50% for tumors larger than 5 cm. In the group of 59 cases studied by Barrow et al.,9 the median OS for tumors 0 to 2.0 cm in size was 80 months, for tumors 2.1 to 4.9 cm 37 months, and for tumors 5.0 cm or more 20 months. In two other studies,7,10 on the other hand, the size of the sarcoma did not correlate with the survival of the patients.
(DFS) rates were 62% and 50%, respectively. Tumor size (less than 5 cm, 5 to 10 cm, or larger than 10 cm) was significantly related to 10-year DFS. Another series of 25 women with primary breast sarcomas and a mean follow-up of 10.5 years was reported by Adem et al.8 Local recurrence occurred in 11 patients after a mean follow-up of 15 months, and 10 patients (40%) had systemic metastases. The 5-year DFS was 90% for tumors 5 cm or less and 50% for tumors larger than 5 cm. In the group of 59 cases studied by Barrow et al.,9 the median OS for tumors 0 to 2.0 cm in size was 80 months, for tumors 2.1 to 4.9 cm 37 months, and for tumors 5.0 cm or more 20 months. In two other studies,7,10 on the other hand, the size of the sarcoma did not correlate with the survival of the patients.
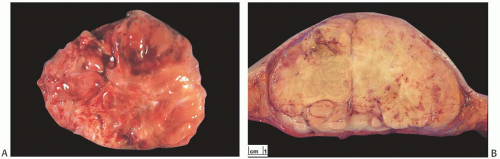 FIG. 39.1. Sarcoma, gross appearance. A: This tumor had areas of osteosarcoma and liposarcoma. B: Fibrosarcoma in a 42-year-old woman replaces much of the breast parenchyma in this mastectomy specimen. Necrosis imparts a pale yellow color centrally. Viable tumor forms a beige rim to the mass and deep secondary nodules [Courtesy of Hyunee Kim, MD). |
Although grading of mammary sarcomas is prognostically important for angiosarcoma, its significance for other types of sarcoma is unclear. Bousquet et al.7 and Zelek et al.,1 for example, found it significant for certain measures of survival, but three other investigations did not demonstrate prognostic significance for the grade of the sarcoma.8,10,11 Hemangiopericytoma represents a special case, since all of these tumors reported thus far have pursued a benign clinical course regardless of size or histologic features.
The adequacy of the definitive treatment represents an influential factor in determining survival. If disease remained after the completion of initial treatment, the 10-year probability of local control and of DFS was 0% for both parameters in one study.7 Several other studies9,10,11,12 detected an adverse effect of positive margins on the chance of survival.
Complete excision of the sarcoma represents the crucial aspect of treatment of mammary sarcomas. The surgical procedure can consist of either a mastectomy or an excision depending on the size of the mass and other surgical and personal considerations. Five studies7,9,10,11,13 have failed to demonstrate a relationship between the type of primary surgical procedure and the survival of the patient. Contemporary multimodal approaches, including irradiation and chemotherapy, may reduce the frequency of local and systemic recurrence in somatic sarcomas, but the results to date are inconclusive.7,9,10,11,13,14,15
Axillary lymph node (ALN) metastases are exceedingly uncommon at the time of primary therapy. In the series of Bousquet et al.,7 metastatic sarcoma was found in 4 of 44 (9%) patients who underwent lymph node evaluation; three of these patients had angiosarcomas. Gutman et al.13 reported that “[R]egional lymph node metastases were always and only in the context of disseminated disease.” Blanchard et al.10 discovered axillary metastases in 2 of 22 patients; both had distant metastases. Axillary dissection or sentinel lymph node (SLN) sampling are not ordinarily indicated in the absence of clinically involved lymph nodes.7,8,10,13,16
Radiation-induced sarcomas of the breast have attracted special interest. A publication by Sheth et al.17 lists 124 original articles describing 1,831 cases of radiation-induced mammary sarcomas. The authors did not analyze the reported histologic features of the sarcomas, but they did note that both the size and the grade of the tumor had prognostic significance. On the basis of the accumulated published data, the authors concluded that “surgery with widely negative margins remains the primary treatment of [radiationinduced sarcomas]. Unfortunately, the role of adjuvant and neoadjuvant chemotherapy remains uncertain.” These conclusions would seem to indicate that neither the nature nor the treatment of radiation-induced sarcomas of the breast differs significantly from those of spontaneously occurring mammary sarcomas.
STROMAL SARCOMA
The term “stromal sarcoma” was introduced in 1962 to describe 25 primary mammary sarcomas that did not qualify for a diagnosis of MPT or angiosarcoma.18 The histologic appearance included “fibrous, myxoid, and fatty patterns.” A liposarcomatous component was identified in six tumors, and others had elements that resembled leiomyosarcoma or
“neurosarcoma.” None of the tumors exhibited osseous or rhabdomyosarcomatous differentiation. The authors concluded that the neoplasms had a common “basic stromallike structure” composed of elongated cells with an “off center” nucleus and, on this basis, chose the term “stromal sarcoma.” The ages of 25 patients ranged from 25 to 64 years, averaging 48 years. All but one were women. Among 15 patients treated by local excision, 9 developed local recurrences within 5 years, and 5 of these patients died of disease. A sixth patient treated by local excision died of sarcoma without local recurrence. Local recurrence occurred in one patient treated by mastectomy whose tumor initially invaded the chest wall. No patient had ALN metastases. The actuarial 5-year survival rate was 60%. In retrospect, the illustrations and microscopic descriptions suggest that the majority of the tumors included in the report would be classified currently as liposarcoma, MFH, or fibrosarcoma. It is now considered preferable to subclassify mammary sarcomas according to their patterns of growth and differentiation using the same terminology as used for sarcomas at other anatomic sites.19,20
“neurosarcoma.” None of the tumors exhibited osseous or rhabdomyosarcomatous differentiation. The authors concluded that the neoplasms had a common “basic stromallike structure” composed of elongated cells with an “off center” nucleus and, on this basis, chose the term “stromal sarcoma.” The ages of 25 patients ranged from 25 to 64 years, averaging 48 years. All but one were women. Among 15 patients treated by local excision, 9 developed local recurrences within 5 years, and 5 of these patients died of disease. A sixth patient treated by local excision died of sarcoma without local recurrence. Local recurrence occurred in one patient treated by mastectomy whose tumor initially invaded the chest wall. No patient had ALN metastases. The actuarial 5-year survival rate was 60%. In retrospect, the illustrations and microscopic descriptions suggest that the majority of the tumors included in the report would be classified currently as liposarcoma, MFH, or fibrosarcoma. It is now considered preferable to subclassify mammary sarcomas according to their patterns of growth and differentiation using the same terminology as used for sarcomas at other anatomic sites.19,20
Despite the lack of diagnostic specificity, the diagnosis of stromal sarcoma is still sometimes used. The breast is composed of specialized, hormonally responsive stroma localized in lobules and around ducts as well as intervening fibrous, adipose, and other mesenchymal tissues. If the term “stromal sarcoma” were selected to designate any of the mesenchymal neoplasms of the breast, it should be applied to those derived from the hormonally responsive specialized stroma; however, most neoplasms arising from this type of stroma belong to the already well-established category of phyllodes tumor (see Chapter 8). The term “periductal stromal sarcoma” has been used for those tumors derived from specialized stroma that lack the leaf-like growth pattern characteristic of phyllodes tumors21,22,23; however, certain cases reported as periductal stromal sarcomas seem to represent phyllodes tumor with myxoid stroma,23 phyllodes tumor with lipomatous stroma,21 or other rare variants of phyllodes tumors.22 Besides differing in the architecture of the stroma, phyllodes tumor and stromal sarcoma differ in the appearance of the glandular component. The glandular tissue within phyllodes tumors demonstrates evidence of proliferation, whereas the ducts and lobules entrapped within stromal sarcomas do not. Those exceptionally rare tumors in which specialized stromal fibroblasts constitute the only proliferative population can be classified as true stromal sarcomas of the breast24 (Fig. 39.2).
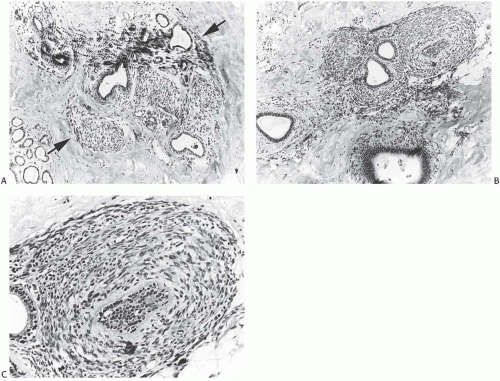 FIG. 39.2. True stromal sarcoma. A: Sarcoma limited to the intralobular stroma between the arrows. The interlobular stroma is normal. B,C: Sarcoma limited to periductal stroma in the same tumor as (A). A,B: Reproduced from Callery CD, Rosen PP, Kinne DW. Sarcoma of the breast. A study of 32 patients with reappraisal of classification and therapy. Ann Surg 1985;201:527-532, with permission.) |
LEIOMYOSARCOMA
Leiomyosarcoma arises in the breast only very rarely and accounts for fewer than 5% of the reported sarcomas of the breast. Fujita et al.25 tabulated clinical features of 46 cases reported as single examples or in small series, Adem et al.26 listed 13 major series of mammary sarcomas, 6 of which contain a total of 9 leiomyosarcomas, and 6 later series of sarcomas of the breast27,28,29,30,31,32 document 15 additional cases. This form of sarcoma probably originates from blood vessels, the smooth muscle of the nipple-areolar complex, or myofibroblasts. A predisposition of smooth muscle in the nipple to give rise to neoplasms is evidenced by reports of leiomyomata33,34,35 and leiomyosarcomas36,37 at this site. Myoid transformation of myoepithelial cells and myofibroblasts are other histogenetic mechanisms.38,39 One leiomyosarcoma may have arisen in an ectopic areola.40
Clinical Presentation
Most patients have been women, but origin in the male breast has been reported.36,41 The age at diagnosis ranges from 1825 to 86 years,42 and the mean age is approximately 53 years. Patients present with a mass measuring from 0.543 to 23 cm44,45 and averaging approximately 5.5 cm. Pain is reported rarely.46 Almost one-half of the tumors are in or near the nipple-areola complex, but any quadrant may be affected. The tumors are circumscribed and firm, and fixation to the skin and ulceration can occur.45,46 A 50-year-old woman who reported a 10-year history of treatment with intermittent low-dose cyclophosphamide for systemic lupus erythematosus developed a 3.2-cm leiomyosarcoma in her left breast.47
Imaging Studies
Mammography reveals a dense, lobulated lesion with a defined border. Calcifications are seen only infrequently.48 In one case,49 pronounced lobulation created the impression of four discrete masses on sonography, and the radiologist described the lesion as “well-circumscribed, oval, and possibly a cluster of fibroadenomas.” Sonograms of another case50 revealed an irregular solid mass with a nonhomogeneous internal echo pattern and an increased anteroposterior dimension and acoustic shadowing. Calcifications were not seen. In one case studied with magnetic resonance imaging (MRI),48 the mass appeared hypointense on T1 images and of heterogeneous intensity on T2 images.
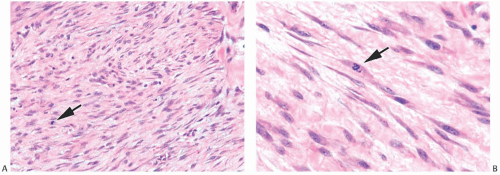 FIG. 39.3. Leiomyosarcoma. A: A moderately cellular tumor composed of fusiform tumor cells with blunt-end nuclei. One mitosis is evident (arrow). B: A mitotic figure is shown [arrow. |
Gross and Microscopic Pathology
Gross examination reveals a circumscribed, firm, lobulated pale tumor. Areas of necrosis may be apparent.
Microscopically, the neoplasm consists of interlacing bundles of fusiform cells with typical blunt-end nuclei characteristic of smooth muscle tumors (Fig. 39.3). Cells with an epithelioid phenotype are variably present. Malignant cytologic features reported in most cases have consisted of nuclear hyperchromasia, pleomorphism (sometimes with multinucleated giant cells), and readily identified mitoses ranging from 251 to 5052 per 10 high-power fields (HPFs), with an average of 12 per 10 HPFs (Fig. 39.4). Focal areas of degeneration are characterized by nuclear pyknosis, necrosis, and lymphocytic infiltration. Areas of hyalinized stromal fibrosis with a pattern that resembles pseudoangiomatous stromal hyperplasia (PASH) may be present (Fig. 39.5). Mammary ducts and lobules, sometimes with proliferative changes, can be incorporated into the neoplasm, particularly at the periphery, a feature that may lead to considering alternative diagnoses such as metaplastic carcinoma and phyllodes tumor. One tumor classified as leiomyosarcoma contained areas of “benign” metaplastic bone and cartilage,53 and another tumor had focal rhabdomyoblastic differentiation.42 Two examples of leiomyosarcoma with osteoclast-like giant cells have been described54,55 (Fig. 39.6). One tumor was classified as an epithelioid leiomyosarcoma.56
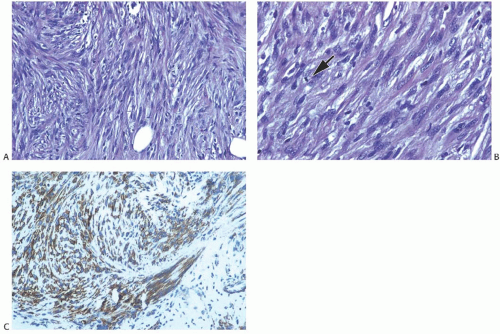 FIG. 39.4. Leiomyosarcoma. A: A highly cellular tumor composed of interlacing fascicles of spindle cells with hyperchromatic pleomorphic nuclei. B: A mitotic figure is shown (arrow). C: Immunoreactivity for SMA. |
Cytology
Fine-needle aspiration (FNA) specimens demonstrate dissociated and poorly cohesive clusters of plump, spindle, or polygonal cells that vary in size and shape.37,46,54,57,58,59 The cells contain moderate amounts of cytoplasm, which can contain small, round, and smooth vacuoles.54 On occasion, the vacuoles appear sufficiently large to create signet ring cells. The nuclei measure 3 to 8 times the size of an erythrocyte, and they usually occupy an eccentric position. They appear pleomorphic and hyperchromatic and they display round, oval, or irregular shapes. The nuclei contain one or more nucleoli and occasional intranuclear cytoplasmic invaginations. Mitotic figures can be identified often. The extent of pleomorphism, hyperchromasia, and mitotic activity in the FNA specimen reflects the histologic appearance of the neoplasm.
 |
Immunohistochemistry
Immunohistochemical staining is usually at least focally positive for desmin, smooth muscle actin (SMA), and vimentin (Figs. 39.4 and 39.6), and reactivity for H-caldesmon46 has been reported. Most examples were negative for cytokeratin (AE1/3, CAM5.2), S-100, and epithelial membrane antigen (EMA), but rare examples have been weakly positive for cytokeratin37 and S-10037,60,61 and focally positive for EMA.60 The precise classification of the tumors with aberrant cytokeratin immunoreactivity remains uncertain. A few examples stained only weakly or not at all for actin, desmin, or SMA.27,42,43,48,56,58,61 Weak reactivity for vimentin has been described.37,61,62 One leiomyosarcoma did not stain for HMB45.63 Weak or absent staining for CD34, CD68,
factor VIII, myoglobin, α1-antichymotrypsin, neuron-specific enolase (NSE), and p53 was reported in isolated cases.42,52,56,57
factor VIII, myoglobin, α1-antichymotrypsin, neuron-specific enolase (NSE), and p53 was reported in isolated cases.42,52,56,57
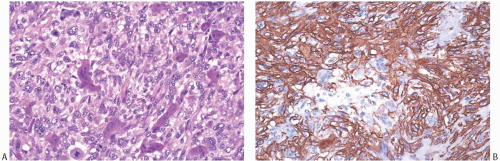 FIG. 39.6. Leiomyosarcoma, osteoclast-like giant cells. A: Osteoclast-like giant cells mingle with epithelioid leiomyosarcoma cells. Mitoses are present in sarcoma cells. B: Strong SMA immunoreactivity is limited to the sarcoma cells. The giant cells were immunoreactive for CD68, a histiocytic marker (not shown), and reactivity for cytokeratin was not present. |
In one case,59 the Ki67 score was 5%, whereas in another,43 the presenting tumor displayed a Ki67 score of 0% and the recurrence a score of 34%. Jun Wei et al.54 reported staining for MIB-1 in 30% of the cells in one tumor.
Electron Microscopy
Electron microscopy has been carried out on just a few cases.36,43,51,56,57,60,64,65,66 The studies revealed spindle cells with nuclei, ribosomes, mitochondria, variably developed rough endoplasmic reticulum, thin basal lamina, and pinocytotic vesicles. The chromatin appeared randomly dispersed, and scattered condensations of chromatin could be seen at the periphery of the nuclear membrane. Myofilaments were described in eight cases.51,56,64,66
Genetic Studies
One tumor was aneuploid.36 Comparative genomic hybridization (CGH) analysis of two leiomyosarcomas detected six chromosomal aberrations in one case and nine aberrations in the other.63 The alterations consisted of losses located at 13q11-q21, 10q23-qter, and 17p and gains at 17p or 1q. Chromosome 13q and 10q deletions were observed in both examples, and these alterations have been seen in approximately 75% of leiomyosarcomas arising in the uterus and deep soft tissues.
Treatment and Prognosis
The publication by Rane et al.67 lists the treatment and followup of most cases published in the English literature. Primary treatment consisted of total mastectomy in approximately 75% of the patients. Twelve patients26,43,49,51,52,54,60,62,64,67,68 underwent excision alone. Of these, seven developed local recurren ces26,43,51,60,64,68 and two died.26,51 Local recurrence on the chest wall after mastectomy has been reported.37,46,69 The likelihood of local recurrence may depend on the amount of seemingly uninvolved surrounding tissue removed during the primary surgical procedure. Fujita et al.25 suggested a margin of 3 cm or more. Axillary nodal metastases have not been reported.
Irradiation has been used in circumstances that did not permit excision with an adequate margin44,45 and to treat recurrences on the chest wall.37,46 Chemotherapy has been administered in just a few cases,26,37,43,46 so its value cannot be assessed.
Among the nearly 50 reported cases of mammary leiomyosarcoma, 6 patients died of the disease.26,29,37,51,56 Outcome has not correlated well with the mitotic rate in the primary tumor, because fatalities occurred in cases with 2 or 3 mitoses per 10 HPFs as well as in tumors with higher mitotic rates. Late recurrences and death from disease 15 and 20 years after initial diagnoses have been reported.51,65
LIPOSARCOMA
This malignant neoplasm may arise from periductal or perilobular stroma in the form of an MPT70,71 or from interlobular stroma to present as a primary stromal sarcoma.70 Liposarcoma arising in PTs is discussed in Chapter 8. Liposarcomas account for approximately 5% of mammary sarcomas reported in the literature.
Clinical Presentation
Patients’ ages range from 1672 to 9073 years at the time of diagnosis with an average age of 49 years. Two male patients have been described.70 The presenting symptom is a mass of variable duration occasionally accompanied by pain.70,74
Two instances of bilateral low-grade liposarcoma have been described,74,75 and the patient described by Lifvendahl76 had several nodules in both breasts. One patient was pregnant at the time of diagnosis,70 and three came to attention during the postpartum period.76,77,78 Radiation-related pleomorphic liposarcoma of the chest wall has been reported 17 months after excision and irradiation79 and 10 years after mastectomy.80
Two instances of bilateral low-grade liposarcoma have been described,74,75 and the patient described by Lifvendahl76 had several nodules in both breasts. One patient was pregnant at the time of diagnosis,70 and three came to attention during the postpartum period.76,77,78 Radiation-related pleomorphic liposarcoma of the chest wall has been reported 17 months after excision and irradiation79 and 10 years after mastectomy.80
On clinical examination, the tumor is typically firm and well circumscribed, but ill-defined lesions have been reported. The overlying skin is usually unaffected, but ulceration was observed associated with a 20-cm tumor81 and another that spanned 28 cm.73
Imaging Studies
The mammographic features of mammary liposarcoma vary. Two examples79,82 appeared well-defined and smoothly outlined, and these features suggested the diagnosis of fibroadenoma (FA). On the other hand, other examples had ill-defined margins,83 and mammography in a patient with bilateral involvement revealed “a bizarre pattern of widespread density in the posterior and axillary region of each breast.”74 Sonography may demonstrate a mass with a complex echo pattern produced by the presence of cystic and solid components77,83 or a lesion suggestive of a benign tumor such as a FA78 or a “fibroadenolipoma.”79 MRI of one tumor revealed a hypodense mass with a distinct border on T1-weighted images, and two tumors appeared hyperdense on T2-weighted scans.83,84
Gross and Microscopic Pathology
The tumors have measured 2 to 40 cm, averaging about 8 cm. Some tumors looked well circumscribed or encapsulated, whereas others appeared multinodular or infiltrative. The tumors have been described as “… greasy, yellow-grey, bulky, circumscribed masses. When sectioned, the tumour appears to bulge from its cut surface. Some areas have a slimy gelatinous appearance; it is from these areas that a thick mucoid secretion may be expressed.”85 The gelatinous areas suggest the presence of a component of myxoid liposarcoma.78 Necrosis and cavitation can occur.73,77,86
The histologic features of liposarcoma in the breast are identical to those of liposarcoma arising in the extremities or trunk. Among published reports, including 25 mammary tumors classified in the manner used for liposarcomas in other sites, 14 (41%) were myxoid (Fig. 39.7), 9 (26%) were well differentiated, 7 (21%) were pleomorphic, and 4 (12%) were poorly differentiated.70,73,74,78,81,82,83,85,86,87,88,89,90,91,92,93,94,95 The well-differentiated liposarcomas include several examples of sclerosing or fibrous liposarcomas.73,82,83 There was no apparent consistent relationship between tumor type, tumor size, and patient age at diagnosis.
Cytology
Specimens obtained by needle aspiration demonstrate variably cellular smears with atypical cells occurring singly and in small groups.78,79,81 The shapes of the cells vary from spindly to round or polygonal. The nuclei appear hyperchromatic and contain coarse chromatin and occasional nucleoli. The pale cytoplasm contains vacuoles of varying size, which indent the nucleus. Oil red O stain demonstrates the presence of lipid in the vacuoles.79 In one case, the vacuolated cells were mistaken for vacuolated neoplastic lobular cells.74 Multinucleate large cells are sometimes seen.81 The presence of a background branching capillary network and myxoid material would suggest the diagnosis of a myxoid liposarcoma.78
Immunohistochemistry
Electron Microscopy
Electron microscopy demonstrates pleomorphic tumor cells with eccentric, large, indented nuclei.81 Ultrastructural study of a well-differentiated liposarcoma96 revealed cells with small amounts of endoplasmic reticulum, rare mitochondria, and membrane-bound cytoplasmic droplets. Large, sometimes indented nuclei sat at the periphery of the cells, and the cell membranes lacked desmosomes. Basal lamina and granular extracellular material surrounded many cells.
Treatment and Prognosis
In many cases, treatment has consisted of simple or radical mastectomy.70 Nandipati et al.77 tabulated the treatment and outcome of many of the reported cases. Wide local excision constituted the primary surgical procedure in seven cases.70,73,79,83,97 ALN metastases have not been reported. With follow-up ranging from less than a year to 20 years, approximately 70% of patients have remained recurrence-free, 6% were alive with systemic recurrence, and 24% died of metastatic liposarcoma.70,74,87,88,89,92,93,94,98 Systemic recurrences and deaths due to disease usually occurred within 2 years of diagnosis and were limited to patients with pleomorphic or high-grade tumors. Tumor size by itself was not predictive of outcome. In isolated cases, systemic chemotherapy and irradiation have been used for palliative purposes.
OSTEOSARCOMA AND CHONDROSARCOMA
Malignant tumors displaying the features of extraskeletal osteosarcomas or chondrosarcomas occasionally arise in the breast. Publications by Silver and Tavassoli99 and Trihia et al.100 list many of the reported cases, and several cases have been published since.84,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116 Several authors point out that most mammary neoplasms with malignant osseous or cartilaginous differentiation are variants of heterologous metaplastic carcinoma or MPTs. Rakha et al.117 adopt a more
extreme position and suggest that essentially all mammary malignancies showing osseous or chondroid features represent either matrix-producing metaplastic carcinomas or phyllodes tumors with massive stromal overgrowth. Whatever the pathogenesis of these malignant tumors, a body of literature describes their clinical and pathologic features.
extreme position and suggest that essentially all mammary malignancies showing osseous or chondroid features represent either matrix-producing metaplastic carcinomas or phyllodes tumors with massive stromal overgrowth. Whatever the pathogenesis of these malignant tumors, a body of literature describes their clinical and pathologic features.
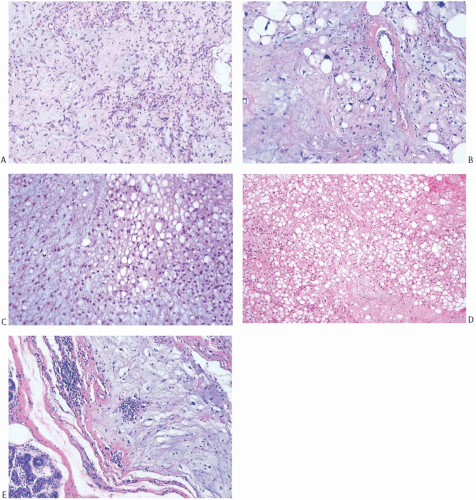 FIG. 39.7. Liposarcoma. A,B: The characteristic stroma and vascular pattern of a myxoid tumor are shown. C: Most of the cells have small, round nuclei in this tumor with myxoid and lipocytic elements. D: Entirely lipocytic growth. E: Myxoid liposarcoma invading breast parenchyma. |
Clinical Presentation
The ages of the patients range from 16112 to 96 years.102 The mean age in the largest series of osteosarcomas was 64.2 years.” Origin in the male breast has been described rarely.99 The presenting symptom is a mass typically described as circumscribed and freely movable. A minority of the tumors are irregular or multinodular. Fixation to the skin or the chest wall and ulceration of the skin occur in a minority of cases.118 The ulceration may result from stretching of the skin rather than from infiltration by the malignant cells.119 One osteosarcoma arose in the breast of a woman 9 years after excision and irradiation of a breast carcinoma in the same breast,99 and radiation-associated postmastectomy osteosarcoma and chondrosarcoma of the chest wall have been reported.120,121
Imaging Studies
Mammography reveals a dense mass, which can appear either well-defined102,108,109,122 or ill-defined.104 The mammographic appearance sometimes suggests the diagnosis of FA.99,105,113,123 Tumors in which the osteosarcomatous component dominates appear heavily calcified. The patterns of the calcifications seen in these tumors have varied. Certain calcifications have been described as “soft,”108,124 and others as “a cluster of crushed stone-like calcifications, which gave the tumor a raw cotton-like appearance.”107 Several authors noted dense central calcifications and finer peripheral ones.109,112 Coussy et al.103 described the mammographic features of one mass as “… containing the following components: a large macrocalcification closely resembling bone, with lobulated borders; a peripheral soft-tissue mass with well-defined borders; and a transition zone between the two that was denser than the soft-tissue mass, had a striated aspect (resembling a perpendicular periosteal hair-on-end reaction) and contained small calcifications.” Tumors of a chondrosarcomatous nature appear hyperdense,122,125 and they may contain calcifications.125 The sarcoma may appear hypoechoic by sonography. One example had heterogeneous echogenicity with an anterior halo and posterior shadowing and enhancement,124 and another101 demonstrated “a densely shadowing mass, compatible with the extensive calcification seen on mammography. Increased through transmission was also present.” Sonograms of other cases showed complex masses with cystic and solid regions and posterior shadowing.104,107,109,113,125 Marked surrounding vascularity was noted on Doppler evaluation in one case.101 These tumors may be positive on a technetium-99 scan.103,109,111,126,127,128 Computed tomography (CT) scans disclose the mass,112 and findings suggestive of hemorrhage, necrosis, and bone formation can be seen on MRI studies in certain cases.103,109
Gross and Microscopic Pathology
The excised tumors have measured 5 to 25 cm in diameter, with an average size of 10 cm. A well-defined border is described in most cases. The cut surface usually has a variegated appearance with areas of softening or gelatinous degeneration and foci of necrosis distributed in firm gray or white tissue. A gritty sensation is encountered when cutting the tumor in areas of ossification. In addition, gross calcification may be visible and palpable.129
Histologic examination reveals a spectrum of microscopic patterns (Fig. 39.8). The tumors have in common a prominent component of high-grade spindle cell sarcoma with a variable mitotic rate. Mitotic counts as high as 52 per 10 HPFs have been noted.101 Tumors with chondroid differentiation alone are less frequent than those with osseous differentiation in which there may be a chondroid element. Most of the former tumors have been reported as single case reports.118,119,122,125,130,131,132,133,134 Beltaos and Banerjee135 described two such cases, and Kennedy and Biggart136 included one example in their group of mammary sarcomas. Multinucleate osteoclastic giant cells are usually present in areas of bone formation.115 Rarely, giant cells constitute a conspicuous element, and they may be associated with hemorrhagic cysts with a telangiectatic appearance.116,137
Cytology
The findings in an FNA specimen depend on the composition of the lesion.100,123,124,137,138,139 The smears usually appear cellular, but hypocellular aspirates sometimes occur. Large atypical cells lie singly and in loose clusters. The cells appear pleomorphic and possess one or two oval- or spindleshaped nuclei, coarse chromatin, prominent nucleoli, and moderate amounts of dense cytoplasm, which was described as “filamentous” in one report.139 Atypical mitotic figures can be seen, and osteoclast-like giant cells may be present. Small-or medium-sized plaques of osteoid, which stains redpurple with the May-Grünwald-Giemsa stain, may be seen in the background. The plaques have round, polyhedral, or bizarre shapes and are sometimes surrounded by malignant cells. If the predominant cellular elements consist of spindle and giant cells, the distinction between a primary sarcoma, an MPT, and a metaplastic carcinoma cannot be made with certainty.
Immunohistochemistry
Immunohistochemical staining for cytokeratins, myoepithelial proteins, and molecules found in muscle cells are negative. Lack of reactivity for cytokeratin and myoepithelial markers is essential to rule out an epithelial component and thereby to exclude the diagnosis of metaplastic carcinoma. Areas with cartilaginous differentiation can be immunoreactive for EMA,99,102,140 and reactivity for S-100 is sometimes found in cartilaginous lesions.99,102,130,131,132,139,140 Staining for SMA has varied in the few cases tested.102,103,131,132 Most tumors in the series of Silver and Tavassoli99 stained for α1-antichymotrypsin and α1-antitrypsin. The cells in isolated cases have failed to stain for polyclonal carcinoembryogenic antigen (CEA), c-kit, CD57, and CD34.102,107,131 Osteoclastic giant cells were immunoreactive for KP-1 (CD68), a marker of histiocytic differentiation.99,103,139,141 A Ki67 score of 20% was reported in one case.103 None of the cases studied have expressed ER, PR, or HER2.99,101,102,103,105,106,108,119,122
Electron Microscopy
The electron microscopic features of six osteosarcomas99,129,142 and one chondrosarcoma130 have been reported. The malignant osteoblasts appeared elongated and possessed conspicuous and often dilated rough endoplasmic reticulum, conspicuous perinuclear Golgi, eccentric nuclei, and large nucleoli. Large osteoclast-like cells contained abundant mitochondria and nondilated rough endoplasmic reticulum. The cell borders of some of the osteoclast-like cells can appear “ruffled.” Undifferentiated round or oval cells, myofibroblasts, and histiocytes may be present. The osteoid had typical long-spaced collagen fibers and hydroxyapatite crystal “puffs.” Desmosomes have been observed linking undifferentiated cells in two osteosarcomas.99,142
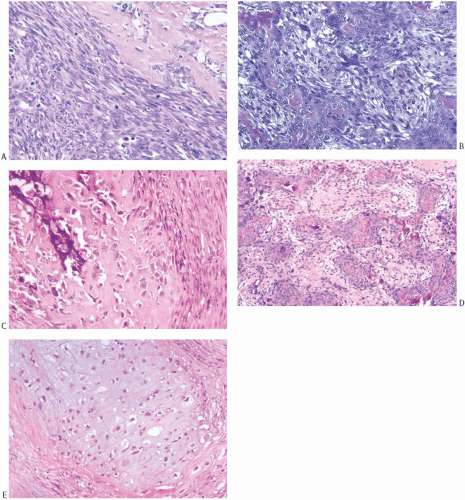 FIG. 39.8. Osteosarcoma and chondrosarcoma. A: Osteoid formation in a high-grade spindle cell sarcoma. B: Patches of osteoid formation in the midst of high-grade spindle cell sarcoma. C: Ossification in osteosarcoma. D: Ossification with an epithelioid appearance in osteosarcoma. E: Chondrosarcoma. |
Treatment and Prognosis
The series of patients studied by Silver and Tavassoli99 provides the most detailed analysis of the treatment and clinical outcome of patients with mammary osteosarcomas. Of the 50 patients in the study group, 18 were treated with limited surgery and 32 with a simple, modified, or radical mastectomy. Local recurrences developed in 67% of patients treated with limited surgery and 11% of those who underwent a mastectomy. Of the eight patients treated with limited surgery who developed local recurrences, seven had positive margins of their surgical specimens. Metastases were not detected in any of the 20 patients who underwent axillary staging. Metastases appeared in 41% of patients, usually within a year from the time of diagnosis. These patients all experienced rapid progression of their disease and died within 20 months. The median survival time was 2 months from the time of detection of the metastases. Using the Kaplan-Meier method, the
probability of OS was 38% at 5 years and 10% at 10 years. Patients with osteosarcomas smaller than 4.6 cm had a higher likelihood for survival than patients with larger tumors, and patients with the fibroblastic type of osteosarcoma had a better prognosis than patients with the osteoclastic or osteoblastic subtypes. The status of the margins of the surgical specimen did not correlate with the likelihood of survival.
probability of OS was 38% at 5 years and 10% at 10 years. Patients with osteosarcomas smaller than 4.6 cm had a higher likelihood for survival than patients with larger tumors, and patients with the fibroblastic type of osteosarcoma had a better prognosis than patients with the osteoclastic or osteoblastic subtypes. The status of the margins of the surgical specimen did not correlate with the likelihood of survival.
Only a few other reports provide follow-up information,100,106,107,110,112,135,137,139,143,144 and the details given support the conclusions reached by Silver and Tavassoli.99 Three publications confirm the absence of ALN metastases in five patients.100,104,106 An 80-year-old woman developed a local recurrence 4 months after a simple mastectomy for osteosarcoma and pulmonary involvement and direct extension into the chest led to her death a few months later.144 Another woman143 developed pulmonary metastases 8 years after the time of diagnosis of a low-grade osteosarcoma; her death ensued 11 months later accompanied by other metastatic deposits.
Irradiation and systemic chemotherapy have been administered in several cases, but the variable nature of these treatments and conditions for which they were used preclude drawing secure conclusions regarding their efficacy.
MFH AND FIBROSARCOMA
Histologic descriptions of spindle cell sarcomas of the breast have not always clearly distinguished between fibrosarcoma and MFH. As noted by Jones et al.,145 “these two tumors have many features in common; thus, classification of some into one or the other group can be arbitrary.” Furthermore, changes in concepts regarding the nature of these tumors as well as their diagnostic criteria make it impossible to determine the nature of many cases reported in the literature. For example, MFH, once considered a specific type of histiocytic neoplasm showing facultative fibroblastic differentiation, now is seen simply as a poorly differentiated sarcoma, which can originate from any of a variety of types of mesenchymal cells. Reflecting this change in thinking, the diagnosis of “MFH” has given way to “undifferentiated pleomorphic sarcoma” when referring to such tumors arising in soft tissues. Pathologists continue to regard fibrosarcoma as specific type of sarcoma derived from cells of a fibroblastic nature; however, the recognition of several specific subtypes of fibrosarcomas has led to reclassification of many of these cases and thereby reduced the number of tumors that remain in the fibrosarcoma category. Bahrami and Folpe146 re-examined 163 tumors of soft tissue classified as fibrosarcomas at the Mayo Clinic between 1960 and 2008 and confirmed the diagnosis in only 26 (16%) cases. The authors reclassified the remaining 137 tumors as 32 examples of MFH, 20 variants of fibrosarcoma, 78 mesenchymal tumors of other types, and 7 nonmesenchymal tumors. The authors concluded that “true [fibrosarcoma] is exceedingly rare … and should be diagnosed with great caution.” Although the writers issued this warning in the context of tumors of soft tissues, it may apply equally well to sarcomas of the breast.
MFH (Undifferentiated Pleomorphic Sarcoma)
For the purposes of the discussion in this chapter, a breast tumor was accepted as MFH if this was the reported diagnosis and the growth pattern was predominantly storiform. Although rare, MFH represents one of the common types of mammary sarcomas. It accounted for 36% of the 240 breast sarcomas in the studies published after 1982 tabulated by Adem et al.147 and 24% of the 25 cases from the Mayo Clinic discussed in this publication. Kijima et al.148 list selected pathologic and clinical features of 31 cases, and 9 series149,150,151,152,153,154,155,156,157 include mention of another 122 examples.
Clinical Presentation
The majority of patients have been women, but low-grade145 and high-grade149,158 MFH of the male breast has been reported. Jeong et al.159 described the case of a 76-year-old man in whom a tumor classified as an “atypical spindle cell lesion” recurred after 1 year as an undifferentiated pleomorphic sarcoma. The age at diagnosis ranges from 24 to 93 years, averaging 52 years. In one series, patients with lowgrade tumors tended to be younger (average age, 46 years) than those with high-grade tumors (average age, 64 years).145 The initial symptom is a mass, which may be located in any portion of the breast. Symptomatic intervals from 1 month to 17 years have been reported.145 The tumors are usually solitary, but patients with multiple tumors have been described.145,160 The overlying skin can exhibit dimpling, induration, ecchymosis, or ulceration.160,161,162 Antecedent trauma is occasionally reported.145
A number of case reports suggest an association between irradiation for breast carcinoma and the development of MFH.163,164 In most of these instances, the sarcoma arose in the chest wall after mastectomy165,166,167,168 or in sites of nodal irradiation such as the axilla.166,169,170 Several patients with breast carcinoma treated with breast conservation and irradiation have developed MFH in the conserved breast. In one case, the sarcoma appeared 2 years after excision and radiotherapy171; in another case,172 the interval between irradiation and the appearance of the sarcoma was 52 months. In one series,166 an interval as long as 7 years between irradiation and the diagnosis of mammary sarcoma was reported.
Gross and Microscopic Pathology
The tumors have measured from 1.0 to 20 cm in size,173 averaging 7.5 cm. In a patient with multiple nodules, the two largest were both 7.0 cm.160 The mass may have a circumscribed or an ill-defined border. The neoplasm is fleshy, firm, or hard and composed of gray, tan, or white tissue. Hemorrhage, necrosis, mucoid change, and calcification are infrequent.
The microscopic hallmark of MFH is the storiform growth pattern in which the spindle cells are arranged in a pin-wheel pattern (Fig. 39.9). Capillaries or small blood vessels may be found at the center of the storiform complex. Giant cells, usually with multiple nuclei, myxoid change, and a chronic inflammatory cell infiltrate, are variably present
(Fig. 39.10). High-grade lesions are characterized by easily identified mitoses, generally numbering more than 3 per HPF, prominent cellular pleomorphism, and necrosis. Lowgrade tumors have little mitotic activity, as well as minimal pleomorphism and necrosis. Cellularity alone is not a reliable criterion for distinguishing between low- and highgrade MFHs of the breast.
(Fig. 39.10). High-grade lesions are characterized by easily identified mitoses, generally numbering more than 3 per HPF, prominent cellular pleomorphism, and necrosis. Lowgrade tumors have little mitotic activity, as well as minimal pleomorphism and necrosis. Cellularity alone is not a reliable criterion for distinguishing between low- and highgrade MFHs of the breast.
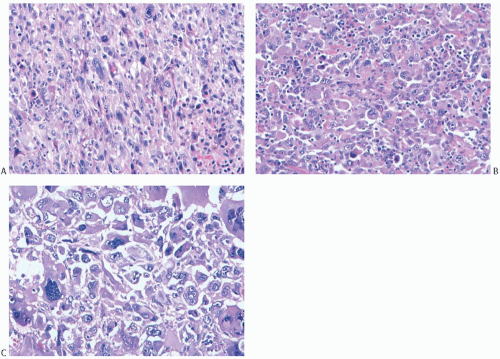 FIG. 39.9. Malignant fibrous histiocytoma. A,B: The characteristic storiform pattern is shown. |
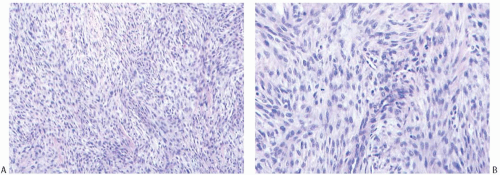 FIG. 39.10. Malignant fibrous histiocytoma. All images are from a single tumor. A: Epithelioid and spindle cells. B: Epithelioid cells with lymphocytes and plasma cells are present. C: Bizarre giant cells formed in a degenerated part of the tumor. |
Cytology
Aspiration cytology yields cellular smears displaying spindle cells, giant cells, and conspicuous capillaries with fragments of collagen in the background.174,175,176 The tumor cells have large, pleomorphic nuclei with irregular nuclear membranes and vacuolated cytoplasm. Both intact cells and naked
nuclei can be seen. Mitotic figures are often numerous, and atypical mitotic figures can be found. Multinucleate tumor cells may be evident. The tumor cells can contain engulfed erythrocytes.176
nuclei can be seen. Mitotic figures are often numerous, and atypical mitotic figures can be found. Multinucleate tumor cells may be evident. The tumor cells can contain engulfed erythrocytes.176
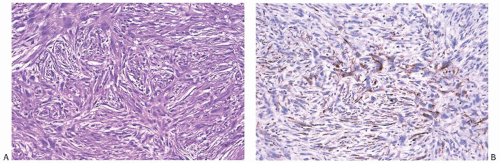 FIG. 39.11. Malignant fibrous histiocytoma. A: A low-grade tumor from a 22-year-old woman. Mitotic figures were infrequent. B: The tumor displayed focal immunoreactivity for CK7 (shown) and vimentin but not for actin. |
Immunohistochemistry
Immunohistochemical stains are not specific. The malignant cells are reactive for vimentin, occasionally for actin,145 and rarely for cytokeratin (Fig. 39.11). All pathologic and clinical features must be given careful consideration when a tumor with storiform growth displays cytokeratin reactivity, since these lesions are usually metaplastic carcinomas, but aberrant cytokeratin expression is present in sarcomas rarely. Immunoreactivity for α1-antitrypsin has been reported in MFH of the breast156 and MFH arising in other organs.177,178
Electron Microscopy
Several publications described the ultrastructural features of MFH.160,167,168,175 The tumors contain a mixture of cell types. Fibroblast-like cells and histiocytic cells predominate. The former have spindly or polygonal shapes, oval nuclei, containing abundant euchromatin, frequent nucleoli, rough and smooth endoplasmic reticulum, abundant ribosomes, mitochondria, lysosomes, Golgi apparatus, and a few droplets of fat. Some fibroblast-like cells contained bundles of filaments resembling myofilaments. The histocytic cells have oval or stellate shapes, abundant cytoplasm containing ribosomes, lysosomes, smooth endoplasmic reticulum, and pleomorphic nuclei. Langerhans cell granules have been seen in histiocytic cells,167 and occasional cells show intercellular junctions. Other types of cells present in smaller numbers include those with both fibroblastic and histiocytic features, multinucleate giant cells, and undifferentiated mesenchymal cells. The tumor cells sit in amorphous matrix containing sparse bands of collagen.
Treatment and Prognosis
With few exceptions, treatment has been by mastectomy with or without axillary dissection. A small minority of patients have been managed successfully by local excision alone. Local recurrence has been reported after both mastectomy and local excision. The choice between mastectomy and local excision depends on the individual clinical presentation and must include consideration of the likelihood of obtaining complete excision and a cosmetically satisfactory result. Rare examples of MFH have metastasized to ALNs,160 but ALN dissection is not indicated unless needed to obtain an adequate margin or the clinical findings suggest the presence of nodal metastases.
Recurrence and death due to disease have been reported in approximately 40% of patients. The most frequent sites of metastases are the lungs and bones. Tumors that developed metastases have been high-grade histologically. Local recurrence is not infrequent among low-grade tumors. Systemic recurrences and deaths usually occurred within 3 years and rarely more than 5 years after diagnoses.145,148,173,175
FIBROSARCOMA
The series of mammary sarcomas listed by Adem et al.147 include 94 tumors said to represent fibrosarcomas, and a few case reports and small series describe or mention several other sarcomas classified as mammary fibrosarcomas. Certain of these cases were phyllodes tumors with a fibrosarcomatous pattern,179 fibrosarcomas arising from a dermatofibrosarcoma protuberans (DFSP),180 or myofibroblastic tumors.181 Most were examined without the benefit of the current understanding of the nature of sarcomas and the availability of ancillary studies such as electron microscopy, immunohistochemical staining, and genetic analysis. Because of these limitations, one has reason to question the validity of the diagnoses in nearly all the published cases.
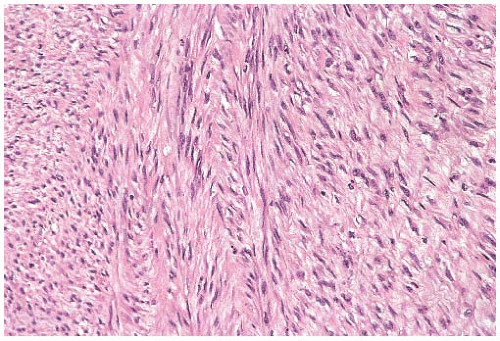 FIG. 39.12. Fibrosarcoma. The tumor consists of spindle cells with elongated nuclei arranged in interwoven bundles. The gross appearance of this tumor is shown in Figure 39.1B. |
Currently proposed histologic criteria for the diagnosis of fibrosarcoma of soft tissue consist of hyperchromatic spindled cells showing no more than moderate pleomorphism, a fascicular, “herringbone” pattern of growth, the presence of interstitial collagen, the absence of morphologic features of all subtypes of fibrosarcomas (myxofibrosarcoma, low-grade fibromyxoid sarcoma, sclerosing epithelioid fibrosarcoma, and fibrosarcoma arising in DFSP), and lack of expression of all markers except vimentin and very minimal SMA.
It seems reasonable to apply these criteria to sarcomas arising in the breast. Thus, mammary sarcomas composed of elongated spindle cells with hyperchromatic spindly nuclei, variably prominent nucleoli, and scant cytoplasm predominantly arranged in broad interdigitating sheets, bands, or fascicles displaying the “herringbone” pattern would be classified as fibrosarcoma (Fig. 39.12). Mitotic figures are almost always evident. The amount of extracellular collagen varies from sparse delicate strands to broad keloidal bands. According to the French system for grading sarcomas (Fédération Nationale des Centres de Lutte Contre le Cancer [FNCLCC]), fibrosarcomas should fall in grade 1 or grade 2 categories. Deviations from these characteristics should prompt consideration of another diagnosis. For example, tumors showing more than a moderate degree of cellular pleomorphism usually merit the diagnosis of undifferentiated pleomorphic sarcoma (MFH). The presence of large areas showing a storiform growth pattern would bring up the diagnosis of fibrosarcoma arising from a DFSP, and the presence of focal adipocytic differentiation would suggest the diagnosis of dedifferentiated liposarcoma. Tumors showing a loosely structured growth pattern without the typical “herringbone” arrangement or showing foci of osteoid formation without osteoblastic differentiation (Fig. 39.13) may represent a specific subtype of fibrosarcoma such as low-grade fibromyxoid sarcoma or myxofibrosarcoma. Finally, detection of proteins characteristic of stromal cells other than fibroblasts would exclude a sarcoma from the fibrosarcoma category. Staining for CD34, for instance, would provoke consideration of the diagnosis of fibrosarcoma arising from either DFSP or solitary fibrous tumor.
One must not forget that other types of sarcomas such as synovial sarcoma, malignant peripheral nerve sheath tumor, solitary fibrous tumor, rhabdomyosarcoma, angiosarcoma, and epithelioid sarcoma and epithelial malignancies such as melanoma and spindle cell carcinoma sometimes display regions resembling fibrosarcomas. Immunohistochemical staining and testing for genetic alterations should allow one to exclude these possibilities in problematic cases.
These considerations suggest that one should probably consider the diagnosis of fibrosarcoma only after excluding all other possibilities. The limited information provided in published cases of mammary fibrosarcomas does not allow one to evaluate the nature of the sarcomas thoroughly; consequently, the diagnosis of fibrosarcoma remains open to question in almost every instance. The case reported by Lee et al.182 may represent a bona fide mammary fibrosarcoma. A 47-year-old woman sought medical attention because of a 10-day history of a 3-cm painless breast mass. Radiologic imaging demonstrated the mass but did not allow for a specific diagnosis. The resected specimen displayed a “firm, fleshy, well-circumscribed, round mass that was gray-white.” Uniform fusiform or spindle-shaped cells with scant cytoplasm and slightly to moderately atypical nuclei embedded in a scant collagenous matrix composed the mass. The mitotic rate was as high as 10 per HPF. The malignant cells failed to stain for cytokeratin, SMA, S-100, and EMA. The patient’s treatment consisted of surgical excision, and she remained free of disease 10 months later.
Postirradiation fibrosarcomas affecting women treated for breast carcinoma have been reported. Most have developed in the chest wall after mastectomy, but rare examples of parenchymal origin after breast conservation and radiotherapy have been reported.183 The authors of the latter publication183 cited several reports of radiation-associated fibrosarcomas of the breast, but they cautioned that “some of these tumors would be classified differently today.”
The lack of well-documented cases of fibrosarcoma makes it impossible to make secure statements about the clinical behavior and optimal treatment of this type of sarcoma. Lacking well-founded information, physicians should probably evaluate and treat patients with fibrosarcomas according to the principles used for other types of mammary sarcomas.
RHABDOMYOSARCOMA
Primary rhabdomyosarcoma of the breast is a very uncommon and poorly characterized neoplasm. Most tumors in the breast with rhabdomyosarcomatous features are variants of metaplastic carcinoma, or MPT,184 or metastases from nonmammary primary sites.185,186,187,188 Evidence for origin in the breast parenchyma or for myomatous differentiation is questionable in most published cases. For example, three rhabdomyosarcomas classified as mammary sarcomas in one report189 “superficially infiltrated skeletal muscle but the tumors were centered in the breast.” Striations were identified microscopically in only one of the three neoplasms. The
other two tumors had “racquet” and “strap” cells with peripherally placed nuclei. Two of the tumors recurred locally with chest wall invasion in one case. Striations were also absent from another sarcoma, which had “strap” and “racquet” cells.184 None of these tumors was examined by electron microscopy, and the reports predate the general availability of immunohistochemistry (IHC).
other two tumors had “racquet” and “strap” cells with peripherally placed nuclei. Two of the tumors recurred locally with chest wall invasion in one case. Striations were also absent from another sarcoma, which had “strap” and “racquet” cells.184 None of these tumors was examined by electron microscopy, and the reports predate the general availability of immunohistochemistry (IHC).
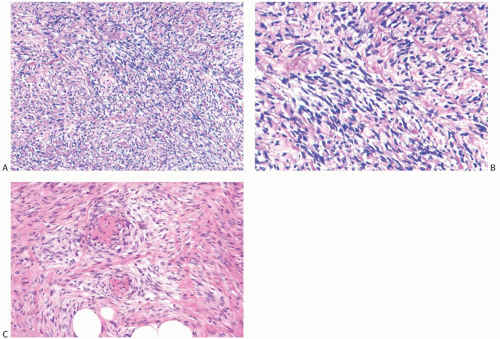 FIG. 39.13. Fibrosarcoma, unusual findings. A,B: Loosely organized groups of spindle cells are present in the collagenous stroma. C: The presence of focal ossification suggests that the tumor may represent a subtype of fibrosarcoma. |
Evans190 described a 4 × 6 × 12 cm circumscribed tumor in the upper outer quadrant of a 41-year-old woman as a mammary rhabdomyosarcoma. “Primitive” cross striations were detected by special stains. The report predated the availability of IHC, and electron microscopy needed to confirm myogenous origin. Recurrence in the muscles of the ipsilateral upper arm, which occurred 32 months after mastectomy, was treated by forequarter amputation. The patient was alive 4 years after diagnosis with recurrent tumor in the supraclavicular region.
A mammary neoplasm with well-documented rhabdomyosarcomatous differentiation was reported by Woodard et al.191 The tumor was a 5-cm mass in the central part of the breast of a 16-year-old girl. The patient remained well 11 years after mastectomy. Microscopic examination revealed rhabdomyosarcoma and a 2.3-cm FA “immediately adjacent without capsular separation.” The FA was described as having “cellpoor stroma.” Cross striations were seen in routine sections, and rhabdomyosarcomatous differentiation was confirmed by electron microscopy. In this instance, origin from the associated fibroepithelial neoplasm cannot be excluded.
Primary Rhabdomyosarcoma
A few primary sarcomas situated in the breast may represent genuine primary mammary rhabdomyosarcomas. The largest series comes from a group of more than 3,500 patients with rhabdomyosarcoma enrolled in the Intergroup Rhabdomyosarcoma Study.188 Within this cohort were seven young women with apparently primary mammary rhabdomyosarcomas; they constitute less than 0.2% of the entire group. Details of the pathologic evaluation were not described in the publication. Nine other reports192,193,194,195,196,197,198,199,200 give details of 10 additional cases. In seven, the rhabdomyomatous nature of the malignant cells was established using immunohistochemical staining for myoglobin, myogenin, or MyoD1. The tumor in one case194 contained the FKHR-PAX3 fusion gene, in another196 the sarcoma contained the typical transcripts for the FKHR-PAX3 fusion gene, and cells in the final case195 demonstrated convincing cross striations. Clinical evaluations did not disclose sites of origin beyond the breasts.
All patients in these reports were female and 13 of 17 were between the ages of 13 and 17 years. Four women were 30, 45 or 46, 46, and 51 years old at the time of diagnosis. The
patients usually recounted the presence of a growing mass. Physical examinations detected nontender masses, usually mobile, without alterations of the overlying skin or nipple. Clinical enlarged ALNs were noted in one patient.199 Sonography of one tumor194 revealed a well-demarcated mass with irregular internal echoes, and CT scanning revealed a huge mass with rich blood flow. In another case,197 an ultrasound study demonstrated a “slightly lobulated, discretely inhomogeneous, hypoechoic tissue density mass.” Using MRI, the mass had high signal intensity on the T2- and nonenhanced T1-weighted images, and dynamic studies showed early enhancement with the formation of a peripheral, strongly vascularized ring. On late dynamic scans, the mass filled completely, but the peripheral hyperintense ring persisted.
patients usually recounted the presence of a growing mass. Physical examinations detected nontender masses, usually mobile, without alterations of the overlying skin or nipple. Clinical enlarged ALNs were noted in one patient.199 Sonography of one tumor194 revealed a well-demarcated mass with irregular internal echoes, and CT scanning revealed a huge mass with rich blood flow. In another case,197 an ultrasound study demonstrated a “slightly lobulated, discretely inhomogeneous, hypoechoic tissue density mass.” Using MRI, the mass had high signal intensity on the T2- and nonenhanced T1-weighted images, and dynamic studies showed early enhancement with the formation of a peripheral, strongly vascularized ring. On late dynamic scans, the mass filled completely, but the peripheral hyperintense ring persisted.
On macroscopic examination, the tumors in the adolescents ranged from 3 to 21 cm in size and those in the adults were smaller (2.5, 3.4, and 4.5 cm). Pathologists described the tumors as round to oval, soft or firm, unencapsulated masses composed of grayish yellow, gray, or white tissue. The presence of necrosis was recorded in one description.195 Microscopical study showed the typical histologic features of rhabdomyosarcoma occurring in conventional sites. The sarcomas were categorized as embryonal in eight of these cases, and alveolar in the remaining nine. The embryonal rhabdomyosarcomas were composed of pleomorphic cells showing varying degrees of skeletal muscle differentiation (Figs. 39.14A




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
