- The rifamycins are semisynthetic antibiotics which have broad-spectrum activity.
- Resistance to the rifamycins arises due to mutations in the rpoB gene, which encodes the β-subunit of RNA polymerase.
- Rifampicin is a potent enzyme inducer and is associated with many clinically significant drug interactions.
- For further information, see Mariani and Maffioli (2009) and Sensi (1983).
Antibacterial rifamycins used clinically are rifampicin (rifampin), rifabutin, rifapentine, and rifaximin; their structures are shown in Figure 2.2.1 and their therapeutic indications are listed in Table 2.2.1.
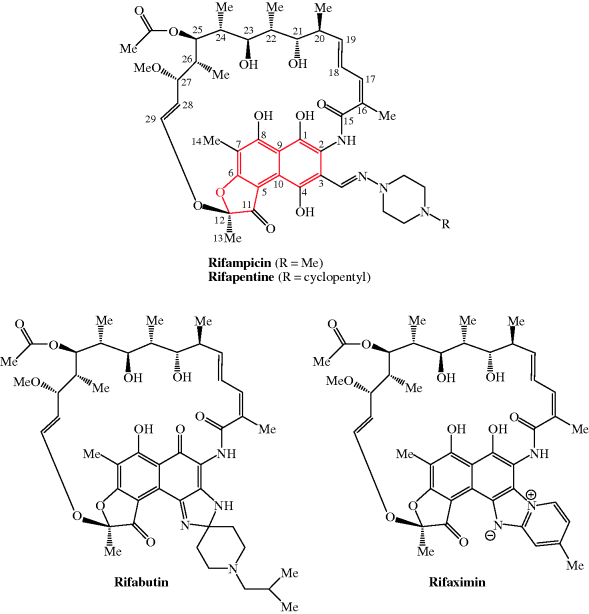
Table 2.2.1 Therapeutic indications for the rifamycin antibacterials.
| Rifamycin antibacterial | Indications |
| Rifampicin (rifampin) | Tuberculosis (TB), leprosy, other mycobacterial infections, meticillin-resistant Staphylococcus aureus, prophylaxis for meningococcal disease |
| Rifabutin | TB, prevention and treatment of M. avium complex (MAC) |
| Rifapentine | TB |
| Rifaximin | Travellers’ diarrhoea |
2.2.1 Discovery
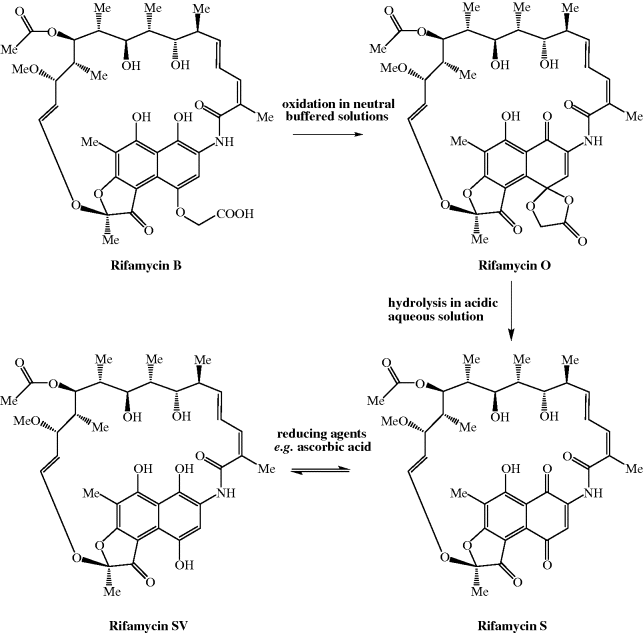
The rifamycins are a class of the ansamycin antibiotics (Mariani and Maffioli, 2009; Floss and Yu, 2005), so called because an aliphatic chain links two nonadjacent carbons of a cyclic system, giving the structure a similarity to the handle of a basket (ansa being the Latin for ‘handle’). In the case of the rifamycins, the aliphatic chain contains 17 atoms, with an ether (C-O) link at one end and an amide (CONH) at the other, while the cyclic system is a naphtho(2,1-b)furan (highlighted in red in the structure of rifamycin in Figure 2.2.1). The rifamycins were first isolated in 1957 from a soil sample, from the beach-side town of St Raphael in France, containing a microorganism which seems to have had some form of identity crisis, having first been classified as Streptomyces mediterranei, then Nocardia mediterranea, and finally Amycolatopsis mediterranei (despite the name changes, there are no prizes for guessing which coast of France St Raphael is on). The antibacterial extract contained a mixture of compounds, with only rifamycin B, the least biologically active component, being isolated in crystalline form. Although rifamycin B had very low activity, it is converted (through a sequence involving spontaneous cyclisation, hydrolysis, and reduction) on standing in solution to rifamycin SV, which has greatly enhanced antibacterial activity and was the first rifamycin introduced into the clinic (Scheme 2.2.1).
2.2.2 Synthesis
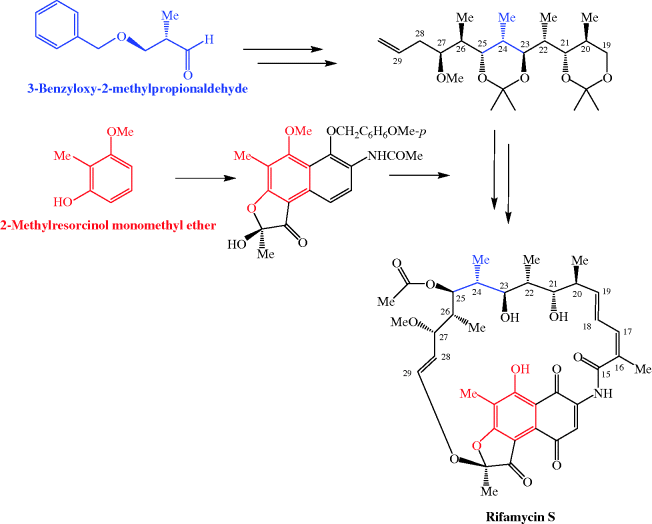
It doesn’t take a long look at the structure of the rifamycins to realise that a total synthesis, involving many functional group interconversions and the stereospecific synthesis of many stereogenic (chiral) centres, would be a supreme challenge. The total synthesis of rifamycin S has attracted the attention of some of the superstars of organic synthesis and the first total synthesis was reported by Kishi and coworkers as early as 1980, in 62 steps, with the ansa chain being derived from 3-benzyloxy-2-methylpropionaldehyde and the aromatic fragment from 2-methyresorcinol monomethyl ether in an elegant synthesis, but, unfortunately, with an overall yield of less than 0.01% (Scheme 2.2.2) (Kishi, 1981 and references cited therein; Paterson and Mansuri, 1985). The production of the rifamycins by such total syntheses is obviously impractical as the quantities obtained are too small, and such a sequence would be too costly, for these drugs ever to have made it to the clinic. How then are the rifamycins obtained? The rifamycins used clinically, like the β-lactams, which we will discuss later, are known as semi-synthetic antibacterial agents as they are prepared by a combination of fermentation and synthesis. Fermentation of a microbial broth produces the complex core structure, containing the ansa ring attached to the naphthofuran ring system, which is then elaborated to give the derivatives used clinically in a series of relatively simple chemical transformations. The key intermediate in the preparation of rifampicin and rifapentine is 3-formylrifamycin SV, which was originally obtained from the decomposition of some of the early, chemically unstable analogues which were synthesised for biological evaluation.
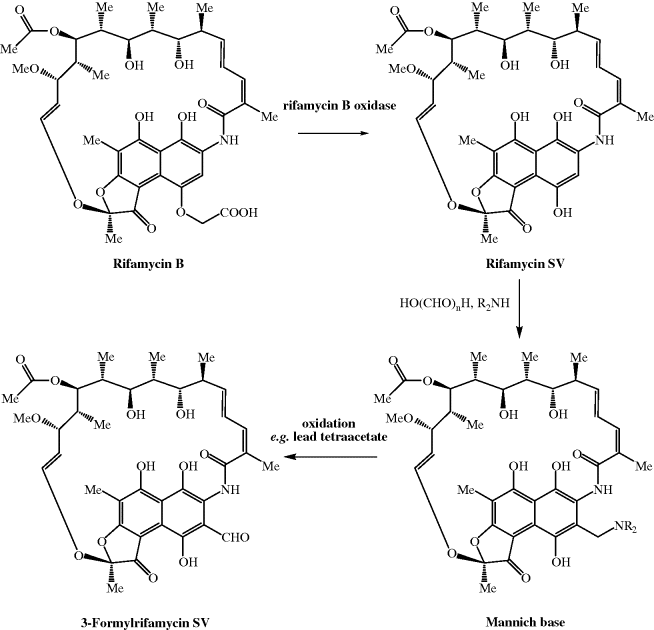
3-Formylrifamycin SV is now obtained by fermentation of A. mediterranei to give rifamycin B, which is converted to rifamycin SV (the chemical conversion gives a low overall yield, so an enzyme-based method has been developed using rifamycin B oxidase) (Lal et al., 1995), and then 3-formylrifamycin SV via the mild oxidation of the Mannich base (Maggi and Sensi, 1967b) formed by reaction with paraformaldehyde and a secondary amine (Scheme 2.2.3). The synthesis of rifampicin and rifapentine then involves the simple condensation of a hydrazine with this aldehyde (Scheme 2.2.4) (Maggi and Sensi, 1967a), a chemical transformation which is very similar to one that many of you will have performed in school/university teaching laboratories: the preparation of yellow/orange crystalline derivatives of aldehydes and ketones through their condensation with Brady’s reagent. As you can imagine, the fermentation of rifamycin-producing microorganisms has been studied extensively, and while rifamycin SV can also be produced by fermentation, the yields are low so that it is more effective to utilise the two-stage route to rifamycin SV.
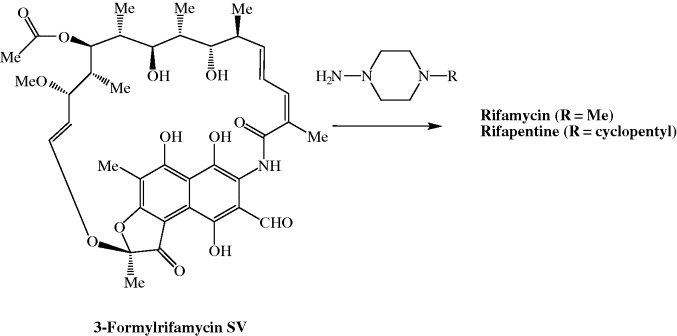
The rifamycins have been the subject of a number of structure–activity relationship (SAR) studies (Sensi and Lancini, 1990) and the key structural requirements for RNAP-inhibitory activity have been identified as:
- Oxygenated substituents at C1 and C8 (OH or C=O) of the aromatic system.
- Free hydroxyls at the C21 and C23 positions with the absolute stereochemical relationship shown in Figure 2.1.11.
Modifications at C3 and/or C4 of the aromatic system, as described above for rifampicin etc., are the most fruitful in terms of increasing the biological activity.
2.2.3 Bioavailability
The rifamycins have different oral bioavailabilities and each is affected differently by co-administration with food. Rifampicin is the fastest and most completely absorbed, with an oral bioavailability of 68%; the presence of food in the GI tract has little effect upon its rate of absorption, but results in a decrease of the maximum serum concentration achieved (Cmax). Rifabutin has lower bioavailability (20%) and the presence of food slows the absorption further. Although poorly studied, the absorption of rifapentine is known to be increased by food, resulting in significantly increased Cmax and AUC values (Burman et al., 2001).
In contrast, rifaximin was specifically designed to remain in the GI tract for the treatment of GI infections, due to its permanently ionised nature, and, as was the intention, is not significantly absorbed after oral administration (Scarpignato and Pelosini, 2005).
The half-lives of the rifamycins in plasma are also quite different and are not dose-dependent: 2–5 hours for rifampicin, 14–18 hours for rifapentine, and 32–67 hours for rifabutin. This property of the latter two allows for once- or twice-weekly administration, enabling directly observed therapy, which we will discuss in Subsection 2.2.6. The systemic rifamycins accumulate in neutrophils, granulocytes, and macrophages, giving these agents an advantage in the treatment of abscesses and bacterial infections involving direct invasion of the immune cells, such as tuberculosis (discussed in Subsection 2.2.6).
The metabolism of the rifamycins is also individual to each, although there are some underlying themes. All three of the absorbed rifamycins are deacetylated at the 25 position by hepatic enzymic action; however, while rifampicin and rifapentine are both hydrolysed to 3-formylrifamycin SV, rifabutin is not metabolised in this way – instead it is hydroxylated by cytochrome P450 isoform 3A (CYP3A). Interestingly, both rifampicin and rifabutin cause autoinduction of their own metabolism, which results in a decreased AUC until steady-state conditions are achieved after about a week. Lastly, all three of these rifamycins cause induction of CYP3A, although to different extents; rifampicin causes the most pronounced induction (and induces other hepatic enzymes too), while rifapentine causes moderate induction and rifabutin’s inductive effect is weak. This CYP3A induction is of particular relevance to drug–drug interactions, of which more will be said later in this section. The rifamycins and their metabolites are largely excreted through the bile into the GI tract and faecally excreted. A relatively small proportion (10–25%) is excreted unchanged in the urine, turning it a red-orange colour1 (Skinner and Blaschke, 1995; Burman et al., 2001; Gumbo et al., 2007).
There is considerable pharmacokinetic inter-individual variability observed in treatment with rifampicin, particularly in cases of comorbidity, such as HIV infection. With these agents, the AUC : MIC ratio is important for effective bactericidal action, but the rifamycins have complex pharmacodynamic parameters and are not described by a simple model. Once- or twice-weekly doses result in a relatively short exposure time to critical concentrations, yet effective antibacterial action can still be achieved. Rifampicin doses of 450 mg/day, rather than the usual 600 mg/day, have been shown to be suboptimal, while higher doses, such as 750 mg/day, are no more effective. Rifapentine, however, shows linear pharmacokinetics over a wide dose range (300–1200 mg) (Burman et al., 2001; Davies and Nuermberger, 2008).
2.2.3.1 Prodrugs
The relatively high logP values of the rifamycins, except for rifaximin, and the acceptable absorption from the GI tract obviate the need for prodrug forms. However, the absorption, stability and antimycobacterial action of rifampicin and isoniazid has been shown to be improved by a polymeric micelle prodrug form of this combination therapy (Silva et al., 2007).
2.2.3.2 Bacterial Uptake of Rifamycins
Rifampicin penetrates well into both Gram positive and Gram negative bacteria, which correlates with its broad spectrum of activity (see Section 2.2.6), and the effects of efflux pumps in reducing intracellular rifamycin concentrations can be overcome by a modest increase in antibacterial concentration (Williams and Piddock, 1998).
2.2.4 Mode of Action and Selectivity
As we shall see in later sections, the rifamycins (well, rifampicin in particular) are now the first-line treatments for Mycobacterium tuberculosis (TB) infections, but they have a very broad spectrum of activity and a high level of activity against Gram positive (e.g. Staphylococcus aureus), Gram negative (e.g. Haemophilus influenzae), and mycobacteria. The rifamycins are bactericidal and their broad spectrum of activity arises from their unique mode of action: the inhibition of bacterial DNA-dependent RNA polymerase (RNAP), a nucleotidyl transferase enzyme that is responsible for the process of transcription (Floss and Yu, 2005; Mariani and Maffioli, 2009). In bacteria, the transcription process can be divided into a number of distinct steps, in which the RNAP holoenzyme binds to duplex promoter DNA to form the RNAP-promoter complex, then a series of conformational changes leads to local unwinding of DNA to expose the transcription start site. RNAP can then initiate transcription, directing the synthesis of short RNA products.
As mentioned in Section 1.1.3.3, RNAP is a complex system, comprising five subunits (α2ββ′ω), each of which has a different function. The α subunits assemble the enzyme and bind regulatory factors, the β subunit contains the polymerase, the β′ subunit binds nonspecifically to DNA, and the ω subunit promotes the assembly of the subunits and constrains the β′ unit. The core structure of RNAP is thought to resemble a crab’s claw – with the active centre on the floor of the cleft between the two ‘pincers’, the β and β′ subunits – and also contains a secondary channel, by which the nucleotide triphosphates access the active centre, and an RNA-exit channel (for a really good interactive tutorial showing the structure of RNAP, see http://www.pingrysmartteam.com/RPo/RPo.htm). Bacterial RNAP contains only these conserved subunits, while eukaryotic RNAP contains these and seven to nine other units (Ebright, 2000).
In bacteria, the transcription of a particular gene requires the binding of a further subunit, a σ factor (a transcription initiation factor), which increases the specificity of RNAP binding to a particular promoter region and so results in the transcription of a particular DNA sequence. Once the assembly process is complete, the holoenzyme (the active form containing all the subunits: α2ββ′ωσ) catalyses the synthesis of RNA, which is complementary to the DNA sequence characterised by the σ factor (Figure 2.2.2) (eukaryotic RNAP also requires the binding of transcription factors).
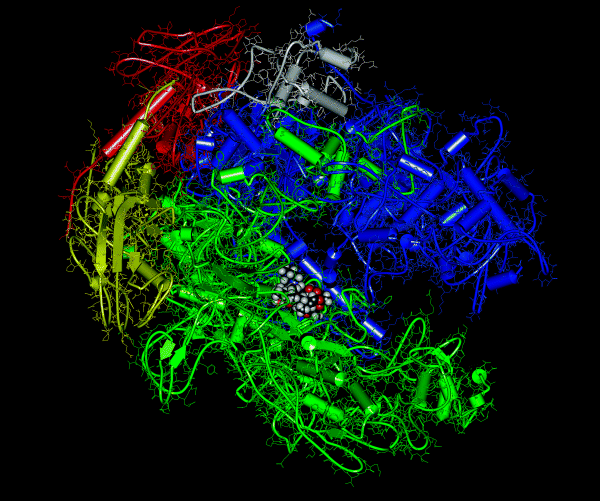
The precise RNAP-binding site of the rifamycins has been the matter of some debate and, despite having been in clinical use since the 1970s, has only just been unequivocally established (famous last words?). One of the mechanisms that has been proposed, the binding of rifamycin to RNAP at a site remote from the catalytic centre, resulting in allosteric modulation of the affinity for the catalytic Mg2+ bound at the active centre (Artsimovitch et al., 2005), was recently discounted (Feklistov et al., 2008). Crystal structures of rifamycin in complex with Thermus aquaticus RNAP, in addition to further mechanistic studies, indicate that rifamycin binds to the β-subunit and in doing so sterically blocks the extension of the RNA chain being formed, after the first or second condensation step, leading to the production of shortened RNA sequences (Zhang et al., 1999). Further support for this mechanism comes from the observation that bacteria are no longer sensitive to the rifamycins once RNA synthesis has produced an oligonucleotide chain.
As with all the antibacterial agents we will be studying, we should ask ourselves why antibacterial agents that target RNAP are selective, as transcription must also take place in eukaryotic cells. As mentioned briefly above, despite structural and functional similarities, the amino acid sequence of the bacterial enzyme differs sufficiently from that of the eukaryotic RNAP, which is also composed of different subunits to those in the prokaryotic RNAP and is present as several variants, each with a different function; for example, eukaryotic RNA polymerase I transcribes ribosomal RNA (rRNA) genes. These differences result in the eukaryotic enzymes being 100–10 000× less sensitive to the rifamycins than their bacterial counterparts (Ho et al., 2009). We learned earlier that the rifamycins are broad-spectrum antibiotics, with activity against mycobacteria, as well as Gram negative and positive organisms, and this results from the fact that the amino acid sequences of their target, the β-subunit of bacterial RNAP, are highly conserved, meaning that rifamycin binding to all bacterial RNAP is equally likely. Differences in bacterial sensitivity, for example between Gram negative and positive species, may thus simply be due to differential permeation of cellular membranes by the rifamycin. For more on mode of action and selectivity, see Hartmann et al. (1985).
2.2.5 Bacterial Resistance
In the previous subsection, we learned about the mode of action of the rifamycins and the reason why they are selective inhibitors of bacterial RNAP: bacterial and eukaryotic RNAP sequences are not highly conserved, resulting in selective toxicity. As we have seen, bacteria respond rapidly to the challenge of an antibiotic agent, and this is particularly true for the rifamycins, with pathogenic bacteria developing resistance at a high rate (typically 10−8–10−9 per bacterium per cell division) (Gillespie, 2002). In order to prevent widespread resistance to these exceedingly useful antibiotics, the rifamycins are thus used in combination therapy, which effectively decreases the chances of resistance developing to any of the agents used in combination, since two, or more, cellular mechanisms are targeted. Use of the rifamycins is also restricted in many countries to the treatment of tuberculosis, or to clinical emergencies, in order to minimise the exposure of bacteria to these agents and so reduce the chances of bacterial resistance developing.
As the rifamycins exhibit their antibacterial effect through binding to the β-subunit of the RNAP, it will come as no surprise that bacterial resistance develops through modifications to the gene encoding this subunit, the rpoB gene, thus diminishing rifamycin-binding affinity for the polymerase. For example, acquired resistance to rifampicin in M. tuberculosis has been shown to arise due to mutations in only 27 codons near the centre of the rpoB gene (Chopra and Brennan, 1998), with more than 70% of mutations found in clinical isolates being due to either His526 to Tyr or Ser531 to Leu (Telenti et al., 1993). These mutations appear to have little effect on the growth of M. tuberculosis, which naturally grows slowly, but result in decreased RNAP affinity for the drug.
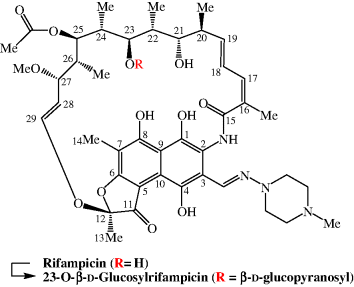
In non-mycobacterial organisms, other resistance mechanisms are encountered, including chemical modification. For example, pathogenic Nocardia species are naturally rifamycin resistant and Nocardia brasiliensis converts rifampicin to its inactive 23-O-β-D-glucosyl derivative (the 21 and 23 hydroxy groups are essential for antibacterial activity) (Figure 2.2.3) (Morisaki et al., 1993). For more on bacterial resistance, see Wehrli (1983), Heym and Cole (1997), and Chopra and Brennan (1998).
2.2.6 Clinical Applications
As mentioned earlier, the target, bacterial RNAP, is well conserved, so the rifamycins have a wide spectrum of activity, with activity against Gram negative bacteria (e.g. Neisseria meningitidis and Haemophilus influenzae), Gram positive bacteria (e.g. Staphylococcus aureus – including, in some cases, MRSA), and mycobacteria (e.g. M. tuberculosis). Despite their spectrum of activity, the clinical applications of the rifamycins are generally limited to the treatment of serious (and sometimes life-threatening) infections in order to minimise the development of bacterial resistance.
2.2.6.1 Tuberculosis
The discovery of the rifamycins helped to transform the treatment of M. tuberculosis by reducing the duration of treatment, increasing cure rates, and decreasing relapse rates for this debilitating disease (Fox et al., 2001). Prior to the rifamycins, treatment of active TB could take up to 18 months, leading to serious problems with patient compliance.
Rifampicin, the first clinically used rifamycin, has activity against both active and latent M. tuberculosis infection and is considered to be a key component of the initial and continuation phases of treatment. Rifampicin should therefore only be excluded from a treatment regimen if the patient has a specific contraindication or if resistance to rifampicin is suspected.
The treatment of tuberculosis is separated into two phases: an initial phase and a continuous phase. The initial phase usually lasts 2 months and is designed to quickly reduce the bacterial population, in order to minimise the possibility of the development of resistance. This is achieved by using four drugs in the initial treatment regimen, usually rifampicin, isoniazid, pyrazinamide, and ethambutol. Resistance to rifampicin normally occurs when treating active M. tuberculosis disease, rather than the latent infection, so it important that the initial phase of treatment is adhered to (Bishai and Chaisson, 1997). The continuous phase starts once the initial phase of treatment is completed, lasts 4 months, and consists of rifampicin and isoniazid.
If the treatment of TB is to be successful, it is critical that the prescribed drug regimen is strictly followed. This is sometimes unrealistic, even with the introduction of combined preparations—for example, Rifater is used in the UK (a preparation combining rifampicin, isoniazid, and pyrazinamide)—as the regimen is often complex, with a high pill burden. In an attempt to overcome this problem, a technique known as directly observed therapy (DOT) has been introduced. DOT involves an appointed person (usually a health worker, community volunteer, or family member) directly supervising patients taking their anti-TB drugs, in order to ensure the medication is taken appropriately. A typical DOT regimen is taken three times per week, while with the unsupervised regimen, the medications are taken daily. Despite the advantages of DOT, a recent Cochrane review concluded that there is insufficient evidence to recommend the routine use of DOT to improve cure rates or treatment completion in people with TB (Volmink and Garner, 2009). Further research is needed to determine in which circumstances DOT would be beneficial, as one small study reported that the use of DOT can actually decrease sucessful treatment outcomes when compared to self-administration (Zwarenstein et al., 1998).
Rifabutin also has activity against M. tuberculosis and, interestingly, its in vitro activity is superior to that of rifampicin (MIC against M. tuberculosis reported as <0.06 mg/mL for rifabutin and 0.15 mg/mL for rifampicin), but rifampicin is still used in the first-line treatment of TB. Despite the positive data for rifabutin, there is still not enough evidence for it to replace rifampicin in the first-line treatment of TB (Davies et al., 2007).
Rifabutin does, in some instances, still have an important role in the treatment of TB and was included in the list agents recommended by the WHO (World Health Organization, 2008). In addition, rifabutin has just been added to the WHO’s list of essential medicines (World Health Organization, 2009b), because it is routinely prescribed (in place of rifampicin) for patients with HIV who are also taking protease inhibitors, since rifampicin is prone to causing significant drug interactions, typically with anti-HIV medication (such as protease inhibitors). Rifabutin has more favourable pharmacokinetic and pharmacodynamic properties and, as it is less likely to cause clinically significant drug interactions, is considered to be a good alternative to rifampicin in patients taking anti-HIV therapy.
Rifabutin may also have a role in the treatment of MDR-TB, which, as discussed in Subsection 2.1.6.5, is becoming a significant clinical problem. Studies have confirmed that some strains of rifampicin-resistant M. tuberculosis are still susceptible to rifabutin, although the evidence suggests that there is a high degree of cross-resistance to rifampicin and rifabutin. For example, one study examining the in vitro activity of rifabutin against M. tuberculosis with rpoB mutations (see Subsection 2.2.5) reported that, of the 41 isolates resistant to rifampicin, 35 were also resistant to rifabutin (Cavusoglu et al., 2004). The WHO guidelines for the management of MDR-TB recommend that a drug should not be included in a treatment regimen if there is a high risk of cross-resistance (World Health Organization, 2008), so that, if a patient contracted MDR-TB resistant to rifampicin, it would not be good practice to include rifabutin in the treatment regimen. If, however, a patient could not tolerate rifampicin (due to side effects or drug interactions, for example) then rifabutin would clearly be an appropriate alternative.
Rifapentine, a long-acting rifamycin, is also used in the management of TB. Rifapentine is available in the USA under the propriety name of Priftin and is licensed for the treatment of pulmonary TB. When rifapentine was first licensed by the FDA in 1998, it was anticipated that its long half-life compared to rifampicin (14–18 hours versus 2–5 hours) would allow less-frequent dosing, or possibly even reduce the duration of treatment. Indeed, a trial published in the Lancet confirmed that once-weekly dosing of rifapentine is effective for the treatment of noncavitary pulmonary TB, although when compared to twice-weekly rifampicin, the rifapentine arm was associated with a higher failure rate (Benator et al., 2002). Furthermore, it has been shown that rifapentine shortens the duration of TB treatment in a murine model when compared to rifampicin-containing regimens (Rosenthal et al., 2007). While these data are encouraging, it is still not clear what role rifapentine has in the international management of TB, and interestingly, it is not yet recommended by the WHO as a first-line therapy (World Health Organization, 2009a). Currently, the American Thoracic Society recommends that rifapentine should be used once weekly (in combination with isoniazid) for the continuous phase of treatment in patients who are HIV sero-negative with non-cavitary, drug-susceptible pulmonary TB (American Thoracic Society, 2003).
2.2.6.2 Meningitis
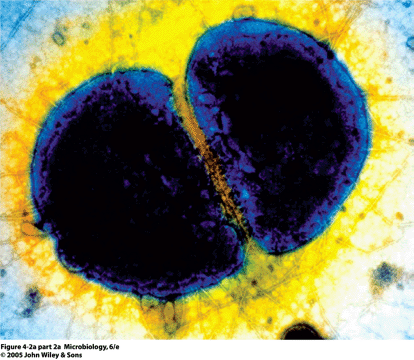
Rifampicin is also used in the prevention of meningitis; a disease characterised by inflammation of the meninges, primarily caused by Neisseria meningitidis – a Gram negative, bean-shaped, diplococcal bacterium (Figure 2.2.4). Meningitis is a serious, often fatal, disease, and even if treated successfully, many patients will sustain severe neurological defects, including loss of feeling in limbs, mental retardation, and paralysis.
Figure 2.2.4 A scanning electron micrograph of Neisseria meningitidis (Reprinted from J. G. Black, Microbiology: Principles and Explorations, 6th Edition, 2005, with permission of John Wiley & Sons.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


