Renin and Angiotensin
The renin–angiotensin system (RAS) participates significantly in the pathophysiology of hypertension, congestive heart failure, myocardial infarction, and diabetic nephropathy.
COMPONENTS OF THE RENIN-ANGIOTENSIN SYSTEM
Angiotensin II (AngII), the most vasoactive angiotensin peptide, participates in blood pressure regulation, aldosterone release, Na+ reabsorption from renal tubules, and electrolyte and fluid homeostasis. AngII is derived from angiotensinogen in two 2 proteolytic steps (Figure 26–1). First, renin, an enzyme released from the juxtaglomerular cells of the kidneys, cleaves the decapeptide angiotensin I (AngI) from the amino terminus of angiotensinogen (renin substrate). Then, angiotensin-converting enzyme (ACE) removes the carboxy-terminal dipeptide of AngI to produce the octapeptide AngII. AngII is an agonist ligand for 2 GPCRs, AT1 and AT2. The RAS includes local (tissue) RAS, alternative pathways for AngII synthesis (ACE-independent), formation of other biologically active angiotensin peptides (AngIII, AngIV, Ang[1–7]), and additional angiotensin-binding receptors (AT1, AT2, AT4, Mas) that participate in cell growth differentiation, hypertrophy, inflammation, fibrosis, and apoptosis.
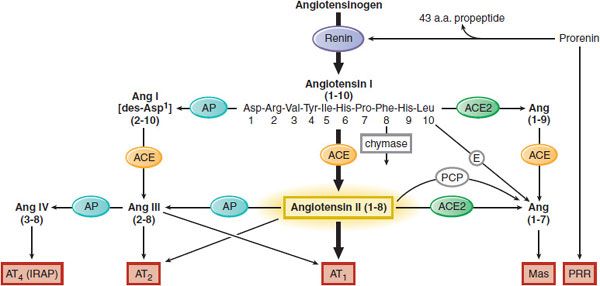
Figure 26–1 Components of the RAS. The heavy arrows show the classical pathway, and the light arrows indicate alternative pathways. ACE, angiotensin-converting enzyme; Ang, angiotensin; AP, aminopeptidase; E, endopeptidases; IRAP, insulin-regulated aminopeptidases; PCP, prolylcarboxylpeptidase; PRR, (pro)renin receptor. Receptors involved: AT1, AT2, Mas, AT4, and PRR. *Exposure of the active site of renin can also occur nonproteolytically; see text and Figure 26–4.
RENIN AND THE PRORENIN/RENIN RECEPTOR. Renin is the major determinant of the rate of AngII production. It is synthesized, stored, and secreted by exocytosis into the renal arterial circulation by the granular juxtaglomerular cells (Figure 26–2) located in the walls of the afferent arterioles that enter the glomeruli. Renin is an aspartyl protease that cleaves the bond between residues 10 and 11 at the amino terminus of angiotensinogen to generate AngI. The active form of renin is a large glycoprotein that is synthesized as a preproenzyme and processed to prorenin. Prorenin may be activated in 2 ways: proteolytically, by proconvertase 1 or cathepsin B that remove 43 amino acids (propeptide) from prorenin’s amino terminus to uncover the active site of renin (Figure 26–3); and nonproteolytically, when prorenin binds to the prorenin/renin receptor (PRR), resulting in conformational changes that unfold the propeptide and expose the active catalytic site of the enzyme. Both renin and prorenin are stored in the juxtaglomerular cells. The concentration of plasma prorenin is ~10x that of the active enzyme. The t1/2 of circulating renin is ~15 min.
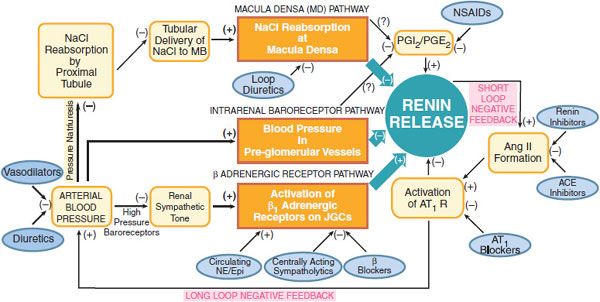
Figure 26–2 Physiological pathways, feedback loops, and pharmacological regulation of the renin-angiotensin system. Schematic portrayal of the three major physiological pathways regulating renin release. See text for details. MD, macula densa; PGI2/PGE2 prostaglandins I2 and E2; NSAIDs, nonsteroidal anti-inflamatory drugs; Ang II, angiotensin II: ACE, angiotensin-converting enzyme. AT1 R, angiotensin subtype 1 receptor; NE/Epi, norepinephrine/epinephrine; JGCs, juxtaglomerular cells.
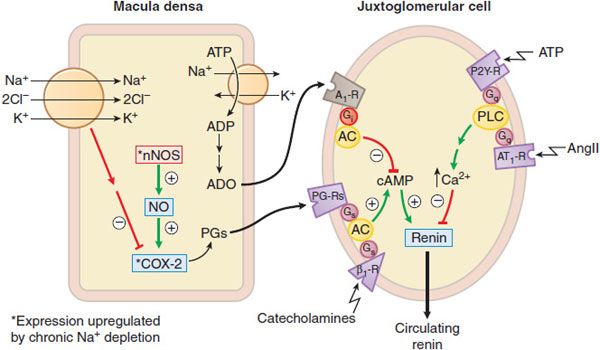
Figure 26–3 Regulation of J-G cell renin release by the macula densa. Mechanisms by which the macula densa regulates renin release. Changes in tubular delivery of NaCl to the macula densa cause appropriate signals to be conveyed to the juxtaglomerular cells. Sodium depletion upregulates nNOS and COX-2 in the macula densa to enhance production of prostaglandins (PGs). PGs and catecholamines stimulate cyclic AMP production and thence renin release from the juxtaglomerular cells. Increased NaCl transport depletes ATP and increases adenosine (ADO) levels. Adenosine diffuses to the juxtaglomerular cells and inhibits cyclic AMP production and renin release via Gi– coupled A1 receptors. Increased NaCl transport in the macula densa augments the efflux of ATP, which may inhibit renin release directly by binding to P2Y receptors and activating the Gq-PLC-IP3-Ca2+ pathway in juxtaglomerular cells. Circulating AngII also inhibits renin release on juxtaglomerular cells via Gq– coupled AT1 receptors.
CONTROL OF RENIN SECRETION. The secretion of renin from juxtaglomerular cells is controlled predominantly by 3 pathways (see Figure 26–2):
1. The macula densa pathway. The macula densa lies adjacent to the juxtaglomerular cells and is composed of specialized columnar epithelial cells in the wall of the cortical thick ascending limb that passes between the afferent and efferent arterioles of the glomerulus. A change in NaCl reabsorption by the macula densa results in the transmission to nearby juxtaglomerular cells of chemical signals that modify renin release. Increases in NaCl flux across the macula densa inhibit renin release, whereas decreases in NaCl flux stimulate renin release.
ATP, adenosine, and prostaglandins modulate the macula densa pathway (Figure 26–3). ATP and adenosine are released when NaCl transport increases: ATP acts on P2Y receptors and adenosine acts via the A1 adenosine receptor to inhibit renin release. Prostaglandins (PGE2, PGI2) are released when NaCl transport decreases, and stimulate renin release through enhancing cyclic AMP formation. Prostaglandin production is stimulated by inducible COX-2 and neuronal NO synthase.
Regulation of the macula densa pathway is more dependent on the luminal concentration of Cl– than Na+. NaCl transport into the macula densa is mediated by the Na+–K+–2CI– symporter (see Figure 26–3), and the half-maximal concentrations of Na+ and Cl– required for transport via this symporter are 2-3 and 40 mEq/L, respectively. Because the luminal concentration of Na+ at the macula densa usually is much greater than the level required for half-maximal transport, physiological variations in luminal Na+ concentrations at the macula densa have little effect on renin release (i.e., the symporter remains saturated with respect to Na+). Conversely, physiological changes in Cl– concentrations (20-60 mEq/L) at the macula densa profoundly affect macula densa–mediated renin release.
2. The intrarenal baroreceptor pathway. Increases and decreases in blood pressure or renal perfusion pressure in the preglomerular vessels inhibit and stimulate renin release, respectively. The immediate stimulus to secretion is believed to be reduced tension within the wall of the afferent arteriole. The release of renal prostaglandins may mediate in part the intrarenal baroreceptor pathway.
3. The β adrenergic receptor pathway is initiated by the release of NE from postganglionic sympathetic nerves. Activation of β1 receptors on juxtaglomerular cells (↑ cyclic AMP) enhances renin secretion.
Increased renin secretion enhances the formation of AngII, which stimulates AT1 receptors on juxtaglomerular cells to inhibit renin release, an effect termed short-loop negative feedback. The inhibition of renin release owing to AngII-induced increases in blood pressure has been termed long-loop negative feedback. AngII increases arterial blood via AT1 receptors; this effect inhibits renin release by: (1) activating high-pressure baroreceptors, thereby reducing renal sympathetic tone; (2) increasing pressure in the preglomerular vessels; and (3) reducing NaCl reabsorption in the proximal tubule (pressure natriuresis), which increases tubular delivery of NaCl to the macula densa.
Renin release is regulated by arterial blood pressure, dietary salt intake, and a number of pharmacological agents (see Figure 26–2). Loop diuretics stimulate renin release by decreasing arterial blood pressure and by blocking the reabsorption of NaCl at the macula densa. Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit prostaglandin synthesis and thereby decrease renin release. ACE inhibitors, angiotensin receptor blockers (ARBs), and renin inhibitors interrupt both the short- and long-loop negative feedback mechanisms and therefore increase renin release. Centrally acting sympatholytic drugs, as well as β adrenergic receptor antagonists, decrease renin secretion by reducing activation of β1 adrenergic receptors on juxtaglomerular cells.
ANGIOTENSINOGEN. AngI is cleaved by renin from the amino terminus of angiotensinogen, an abundant globular protein synthesized mainly by the liver. Angiotensinogen transcripts also are abundant in fat, certain regions of the CNS, and the kidneys. Angiotensinogen synthesis is stimulated by inflammation, insulin, estrogens, glucocorticoids, thyroid hormone, and AngII. During pregnancy, plasma levels of angiotensinogen increase several-fold owing to increased estrogen. Circulating levels of angiotensinogen are approximately equal to the Km of renin for its substrate (~1 μM). Consequently, the rate of AngII synthesis, and therefore blood pressure, can be influenced by changes in angiotensinogen levels. Oral contraceptives containing estrogen increase circulating levels of angiotensinogen and can induce hypertension.
ANGIOTENSIN-CONVERTING ENZYME (ACE, KININASE II, DIPEPTIDYL CARBOXYPEPTIDASE). ACE is an ectoenzyme and glycoprotein that contains 2 homologous domains, each with a catalytic site and a Zn2+-binding region. ACE is rather nonspecific and cleaves dipeptide units from substrates with diverse amino acid sequences. Preferred substrates have only 1 free carboxyl group in the carboxyl-terminal amino acid, and proline must not be the penultimate amino acid; thus, the enzyme does not degrade AngII. ACE is identical to kininase II, the enzyme that inactivates bradykinin and other potent vasodilator peptides. Although slow conversion of AngI to AngII occurs in plasma, the very rapid metabolism that occurs in vivo is due largely to the activity of membrane-bound ACE present on the luminal surface of endothelial cells throughout the vascular system.
The ACE gene contains an insertion/deletion polymorphism in intron 16 that explains 47% of the phenotypic variance in serum ACE levels. The deletion allele, associated with higher levels of serum ACE and increased metabolism of bradykinin, may confer an increased risk of hypertension, cardiac hypertrophy, atherosclerosis, and diabetic nephropathy.
ANGIOTENSIN-CONVERTING ENZYME 2 (ACE2). Human ACE2 contains a single catalytic domain that is 42% identical to the 2 catalytic domains of ACE. ACE2 cleaves 1 amino acid from the carboxyl terminal to convert AngI to Ang(1-9) and AngII to Ang(1-7). AngII is the preferred substrate for ACE2 with 400-fold higher affinity than AngI. ACE2 may serve as a counterregulatory mechanism to oppose the effects of ACE. ACE2 regulates the levels of AngII and limits its effects by converting it to Ang(1-7), which binds to Mas receptors and elicits vasodilator and anti-proliferative responses. ACE2 is not inhibited by ACE inhibitors and has no effect on bradykinin. In animals, reduced expression of ACE2 is associated with hypertension, defects in cardiac contractility, and elevated levels of AngII.
ANGIOTENSIN PEPTIDES. AngI is rapidly converted to AngII. Angiotensin III (AngIII), also called Ang(2–8), can be formed either by the action of aminopeptidase on AngII or by the action of ACE on Ang(2-10). AngII and AngIII cause qualitatively similar effects. AngII and AngIII stimulate aldosterone secretion with equal potency; however, AngIII is only 25% and 10% as potent as AngII in elevating blood pressure and stimulating the adrenal medulla, respectively.
Ang(1-7) is formed by multiple pathways (see Figure 26–1). Ang(1-7) opposes many of the effects of AngII: It induces vasodilation, promotes NO production, potentiates the vasodilatory effects of bradykinin, and inhibits AngII-induced activation of ERK1/2; it has antiangiogenic, antiproliferative, and antithrombotic effects; and is cardioprotective in cardiac ischemia and heart failure. The effects of Ang(1-7) are mediated by a specific Mas receptor. ACE inhibitors increase tissue and plasma levels of Ang(1-7), both because AngI levels are increased and diverted away from AngII formation and because ACE contributes to the plasma clearance of Ang(1-7). AT1 receptor blockade boosts the levels of AngII that is converted to Ang(1-7) by ACE2.
Angiotensin IV (AngIV), also called Ang(3-8), is formed from AngIII through the action of aminopeptidase M and has potent effects on memory and cognition. Central and peripheral actions of AngIV are mediated through specific AT4 receptors identified as insulin-regulated aminopeptidases (IRAPs). AngIV binding to AT4 receptors inhibits the catalytic activity of IRAPs and enables accumulation of various neuropeptides linked to memory potentiation. Other actions include renal vasodilation, natriuresis, neuronal differentiation, hypertrophy, inflammation, and extracellular matrix remodeling. Analogs of angiotensin IV are being developed for their therapeutic potential in cognition in Alzheimer disease or head injury.
LOCAL (TISSUE) RENIN-ANGIOTENSIN SYSTEMS. Many tissues—including the brain, pituitary, blood vessels, heart, kidney, and adrenal gland—express mRNAs for renin, angiotensinogen, and/or ACE, and various cells cultured from these tissues produce renin, angiotensinogen, ACE, and angiotensins I, II, and III. Thus, it appears that local RASs exist independently of the renal/hepatic-based system and may influence vascular, cardiac, and renal function and structure. Activation of (tissue) RAS and local AngII production require the binding of renin or prorenin to the specific (pro)renin receptor (PRR), located on cell surfaces.
The (Pro)Renin Receptor. The PRR is the functional cell surface receptor that binds prorenin and renin with high affinity (KD ~6 and 20 nM, respectively) and specificity. Binding of (pro)renin to PRR enhances the catalytic activity of renin by 4- to 5-fold and induces non-proteolytic activation of prorenin (Figure 26–4). Bound, activated (pro)renin catalyzes the conversion of angiotensinogen to AngI, which can be converted to AngII by ACE located on the cell surface. AngII binds to AT1 receptors and activates intracellular signaling events that regulate cell growth, collagen deposition, fibrosis, inflammation, and apoptosis.
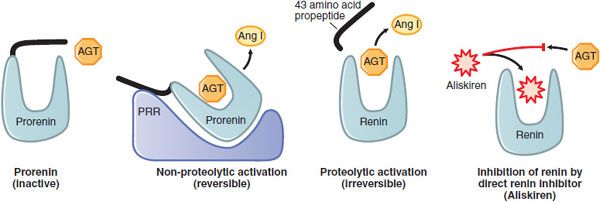
Figure 26–4 Biological activation of prorenin and pharmacological inhibition of renin. Prorenin is inactive; accessibility of angiotensinogen (AGT) to the catalytic site is blocked by the propeptide (black segment). The blocked catalytic site can be activated non-proteolytically by the binding of prorenin to the (pro)renin receptor (PRR) or by proteolytic removal of the propeptide. The competitive renin inhibitor, aliskiren, has a higher affinity (~0.1 μm) for the active site of renin than does AGT (~1 μm).
The binding of (pro)renin to PRR also induces AngII-independent signaling events that include activation of ERK1/2, p38, tyrosine kinases, TGF-β gene expression, and plasminogen activator inhibitor type 1 (PAI-1). These signaling pathways are not blocked by ACE inhibitors or AT1 receptor antagonists and are reported to contribute to fibrosis, nephrosis, and organ damage. PRR is abundant in the heart, brain, eye, adrenals, placenta, adipose tissue, liver, and kidneys.
Prorenin is no longer considered the inactive precursor of renin, as it is capable of activating local RAS and AngII-independent events that may contribute to organ damage. Circulating plasma concentrations of prorenin are 10-fold higher than renin in healthy subjects but are elevated to 100-fold in diabetic patients and are associated with increased risk of nephropathy, renal fibrosis, and retinopathy. The interaction of prorenin with PRR has become a target for therapeutic interventions.
Prorenin and renin also bind to the mannose-6-phosphate receptor (M6P), an insulin-like growth factor II receptor that functions as a clearance receptor. Knockout of the PRR gene is lethal. In humans, mutations in the PRR gene are associated with mental retardation and epilepsy, suggesting an important role in cognition, brain development, and survival.
ALTERNATIVE PATHWAYS FOR ANGIOTENSIN BIOSYNTHESIS. Angiotensinogen may be converted to AngI or directly to AngII by cathepsin G and tonin. Other enzymes that convert AngI to AngII include cathepsin G, chymostatin-sensitive AngII-generating enzyme, and heart chymase.
ANGIOTENSIN RECEPTORS. AngII and AngIII bind to specific GPCRs designated AT1 and AT2. Most of the known biological effects of AngII are mediated by the AT1 receptor. The AT1 receptor gene contains a polymorphism (A-to-C transversion in position 1166) associated with hypertension, hypertrophic cardiomyopathy, and coronary artery vasoconstriction. Preeclampsia is associated with the development of agonistic auto-antibodies against the AT1 receptor.
The functional role of the AT2 receptor is less defined, but may counterbalance many of the effects of the AT1 receptor by having antiproliferative, proapoptotic, vasodilatory, natriuretic, and antihypertensive effects. The AT2 receptor is distributed widely in fetal tissues, but its distribution is more restricted in adults. Expression of AT2 receptor is upregulated in cardiovascular diseases, including heart failure, cardiac fibrosis, and ischemic heart disease; however, the significance of increased AT2 receptor expression is unclear.
The Mas receptor mediates the effects of Ang(1-7), which include vasodilation and antiproliferation. Deletion of the Mas gene in transgenic mice reveals cardiac dysfunction.
The AT4 receptor mediates the effects of AngIV. This receptor is a single transmembrane protein (1025 amino acids) that co-localizes with the glucose transporter GLUT4. AT4 receptors are detectable in a number of tissues, such as heart, vasculature, adrenal cortex, and brain regions processing sensory and motor functions.
ANGIOTENSIN RECEPTOR-EFFECTOR COUPLING. AT1 receptors activate a large array of signal-transduction systems to produce effects that vary with cell type and that are a combination of primary and secondary responses. AT1 receptors couple to several heterotrimeric G proteins, including Gq, G12/13, and Gi. In most cell types, AT1 receptors couple to Gq to activate the PLCβ–IP3–Ca2+ pathway. Secondary to Gq activation, activation of PKC, PLA2, and PLD and eicosanoid production, as well as activation of Ca2+-dependent and MAP kinases and the Ca2+–calmodulin–dependent activation of NOS may occur. Activation of Gi may occur and will reduce the activity of adenylyl cyclase, lowering cellular cyclic AMP content. The βγ subunits of Gi and activation of G12/13 lead to activation of tyrosine kinases and small G proteins such as Rho. Ultimately, the JAK/STAT pathway may be activated and a variety of transcriptional regulatory factors induced. AT1 receptors also stimulate the activity of a membrane-bound NADH/NADPH oxidase that generates reactive oxygen species (ROS). ROS may contribute to biochemical effects (activation of MAP kinase, tyrosine kinase, and phosphatases; inactivation of NO; and expression of monocyte chemoattractant protein-1) and physiological effects (acute effects on renal function, chronic effects on blood pressure, and vascular hypertrophy and inflammation). The presence of other receptors may alter the response to AT1 receptor activation. For example, AT1 receptors heterodimerize with bradykinin B2 receptors, a process that enhances AngII sensitivity in preeclampsia.
Signaling from AT2 receptors is mediated by G protein-dependent and independent pathways. Stimulation of AT2 receptor activates phosphoprotein phosphatases, K+ channels, synthesis of NO and cyclic GMP, bradykinin production, and inhibition of Ca2+ channel functions. AT2 receptors may possess constitutive activity: overexpression of AT2 receptors can induce NO production in vascular smooth muscle cells and hypertrophy in cardiac myocytes through an intrinsic activity of the AT2 receptor independent of AngII binding.
FUNCTIONS AND EFFECTS OF THE RENIN-ANGIOTENSIN SYSTEM
AngII increases total peripheral resistance (TPR) and alters renal function and cardiovascular structure through direct and indirect mechanisms (Figure 26–5).
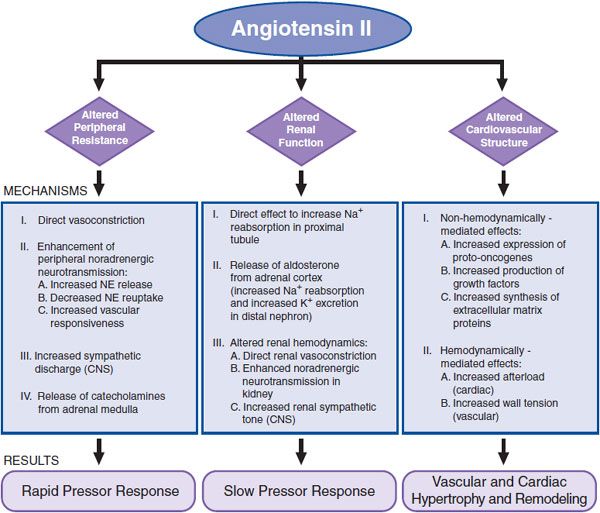
Figure 26–5 Major effects of AngII. NE, norepinephrine.
EFFECTS ON TPR
Direct Vasoconstriction. AngII constricts precapillary arterioles and, to a lesser extent, postcapillary venules by activating AT1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


