CHAPTER 18 Regulation of the Heart and Vasculature
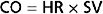
NERVOUS CONTROL OF THE HEART RATE
Parasympathetic tone usually predominates in healthy, resting individuals. When a resting individual is given atropine, a muscarinic receptor antagonist that blocks parasympathetic effects, the heart rate generally increases substantially. If a resting individual is given propranolol, a β-adrenergic receptor antagonist that blocks sympathetic effects, the heart rate usually decreases only slightly (Fig. 18-1). When both divisions of the autonomic nervous system are blocked, the heart rate of young adults averages about 100 beats/min. The rate that prevails after complete autonomic blockade is called the intrinsic heart rate.
Parasympathetic Pathways
The cardiac parasympathetic fibers originate in the medulla oblongata, in cells that lie in the dorsal motor nucleus of the vagus or in the nucleus ambiguus (see Chapter 11). The precise location of the parasympathetic fibers varies among species. In humans, centrifugal vagal fibers pass inferiorly through the neck near the common carotid arteries and then through the mediastinum to synapse with postganglionic vagal cells. These cells are located either on the epicardial surface or within the walls of the heart. Most of the vagal ganglion cells are located in epicardial fat pads near the SA and atrioventicular (AV) nodes.
The right and left vagi are distributed to different cardiac structures. The right vagus nerve affects the SA node predominantly; stimulation of this nerve slows SA nodal firing and can even stop the firing for several seconds. The left vagus nerve mainly inhibits AV conduction tissue to produce various degrees of AV block (see Chapter 16). However, the distribution of the efferent vagal fibers is overlapping such that left vagal stimulation also depresses the SA node and right vagal stimulation impedes AV conduction.
The SA and AV nodes are rich in cholinesterase, an enzyme that rapidly hydrolyzes the neurotransmitter acetylcholine (ACh). The effects of a given vagal stimulus decay very quickly (Fig. 18-2, A) when vagal stimulation is discontinued because ACh is rapidly destroyed. In addition, vagal effects on SA and AV nodal function have a very short latency (≈50 to 100 msec) because the ACh released quickly activates special AChregulated K+ channels (KACh) in the cardiac cells. These channels open quickly because the muscarinic receptor is coupled directly to the KACh channel by a guanine nucleotide–binding protein (Gi). These two features of the vagus nerves—brief latency and rapid decay of the response—permit them to exert beat-by-beat control of SA and AV nodal function.
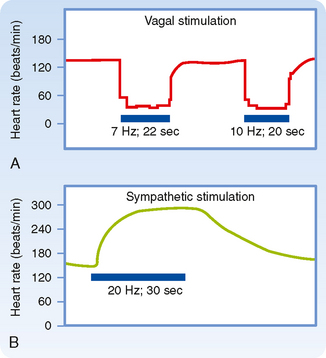
Figure 18-2 Changes in heart rate evoked by stimulation (horizontal bars) of the vagus (A) and sympathetic nerves (B).
(Modified from Warner HR, Cox A: J Appl Physiol 17:349, 1962.)
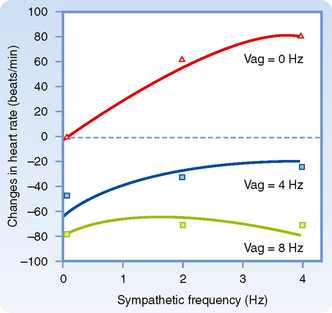
Parasympathetic influences usually predominate over sympathetic effects at the SA node, as shown in Figure 18-3. When the frequency of sympathetic stimulation increases from 0 to 4 Hz, the heart rate increases by about 80 beats/min in the absence of vagal stimulation (Vag = 0 Hz). However, when the vagi are stimulated at 8 Hz, increasing the sympathetic stimulation frequency from 0 to 4 Hz has only a negligible influence on heart rate.
Sympathetic Pathways
The cardiac sympathetic fibers originate in the intermediolateral columns of the upper five or six thoracic and lower one or two cervical segments of the spinal cord (see Chapter 11). These fibers emerge from the spinal column through the white communicating branches and enter the paravertebral chains of ganglia. The preganglionic and postganglionic neurons synapse mainly in the stellate or middle cervical ganglia, depending on the species. In the mediastinum, the postganglionic and preganglionic parasympathetic fibers join to form a complicated plexus of mixed efferent nerves to the heart.
In contrast to abrupt termination of the response after vagal activity, the effects of sympathetic stimulation decay gradually after stimulation is stopped (Fig. 18-2, B). Nerve terminals take up to 70% of the norepinephrine released during sympathetic stimulation; much of the remainder is carried away by the bloodstream. These processes are slow. Furthermore, the facilitatory effects of sympathetic stimulation on the heart attain steady-state values much more slowly than do the inhibitory effects of vagal stimulation. The onset of the cardiac response to sympathetic stimulation begins slowly for two main reasons. First, norepinephrine appears to be released slowly from the sympathetic nerve terminals. Second, the cardiac effects of the neurally released norepinephrine are mediated mainly by a relatively slow second messenger system involving cAMP (see Chapter 3). Hence, sympathetic activity alters the heart rate and AV conduction much more slowly than vagal activity does. Although vagal activity can exert beat-by-beat control of cardiac function, sympathetic activity cannot.
Control by Higher Centers
Stimulation of various brain regions can have significant effects on cardiac rate, rhythm, and contractility (see Chapter 11). In the cerebral cortex, centers that regulate cardiac function are located in the anterior half of the brain, principally in the frontal lobe, the orbital cortex, the motor and premotor cortex, the anterior portion of the temporal lobe, the insula, and the cingulate gyrus. Stimulation of the midline, ventral, and medial nuclei of the thalamus elicits tachycardia. Stimulation of the posterior and posterolateral regions of the hypothalamus can also change the heart rate. Stimuli applied to the H2 fields of Forel in the diencephalon evoke various cardiovascular responses, including tachycardia; these changes resemble those observed during muscular exercise. Undoubtedly, the cortical and diencephalic centers initiate the cardiac reactions that occur during excitement, anxiety, and other emotional states. The hypothalamic centers also initiate the cardiac response to alterations in environmental temperature. Experimentally induced temperature changes in the preoptic anterior hypothalamus alter the heart rate and peripheral resistance.
Baroreceptor Reflex
Sudden changes in arterial blood pressure initiate a reflex that evokes an inverse change in heart rate (Fig. 18-4). Baroreceptors located in the aortic arch and carotid sinuses are responsible for this reflex. The inverse relationship between heart rate and arterial blood pressure is generally most pronounced over an intermediate range of arterial blood pressure. Below this intermediate range, the heart rate maintains a constant, high value; above this pressure range, the heart rate maintains a constant, low value.
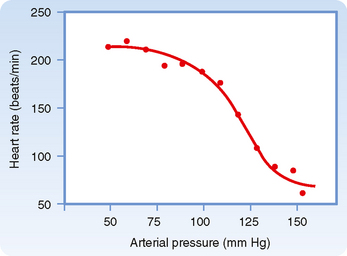
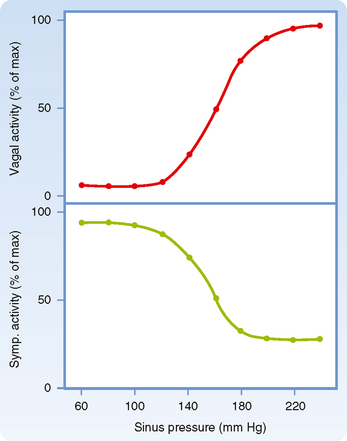
The effects of these changes in carotid sinus pressure on activity in the cardiac autonomic nerves are presented in Figure 18-5, which shows that over an intermediate range of carotid sinus pressure (100 to 180 mm Hg), reciprocal changes are evoked in efferent vagal and sympathetic neural activity. Below this range of carotid sinus pressure, sympathetic activity is intense and vagal activity is virtually absent. Conversely, above the intermediate range of carotid sinus pressure, vagal activity is intense and sympathetic activity is minimal.
Bainbridge Reflex, Atrial Receptors, and Atrial Natriuretic Peptide
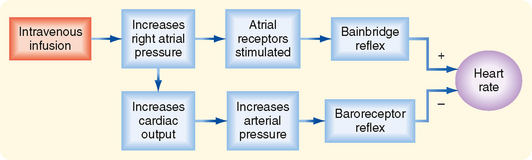
In 1915, Bainbridge reported that infusing blood or saline into dogs accelerated their heart rate. This increase did not seem to be tied to arterial blood pressure because the heart rate rose regardless of whether arterial blood pressure did or did not change. However, Bainbridge also noted that the heart rate increased whenever central venous pressure rose sufficiently to distend the right side of the heart. Bilateral transection of the vagi abolished this response. This is termed the Bainbridge reflex.
Many investigators have confirmed Bainbridge’s observations and have noted that the magnitude and direction of the response depend on the prevailing heart rate. When the heart rate is slow, intravenous infusions usually accelerate the heart. At more rapid heart rates, however, infusions ordinarily slow the heart. What accounts for these different responses? Increases in blood volume not only evoke the so-called Bainbridge reflex but also activate other reflexes (notably the baroreceptor reflex). These other reflexes tend to elicit opposite changes in heart rate. Therefore, changes in heart rate evoked by an alteration in blood volume are the result of these antagonistic reflex effects (Fig. 18-6). Evidently, the Bainbridge reflex predominates over the baroreceptor reflex when blood volume rises, but the baroreceptor reflex prevails over the Bainbridge reflex when blood volume diminishes.
Stimulation of the atrial receptors increases not only the heart rate but also urine volume. Reduced activity in the renal sympathetic nerve fibers may partially account for this diuresis. However, the principal mechanism appears to be a neurally mediated reduction in vasopressin (antidiuretic hormone) secretion by the posterior pituitary gland (see Chapters 34 and 40). Stretch of the atrial walls also releases atrial natriuretic peptide (ANP) from the atria.* ANP, a 28–amino acid peptide, exerts potent diuretic and natriuretic effects on the kidneys (see also Chapter 34) and vasodilator effects on the resistance and capacitance vessels. Thus, ANP is an important regulator of blood volume and blood pressure.
Respiratory Sinus Arrhythmia
Rhythmic variations in heart rate, occurring at the frequency of respiration, are detectable in most individuals and tend to be more pronounced in children. The heart rate typically accelerates during inspiration and decelerates during expiration (Fig. 18-7).
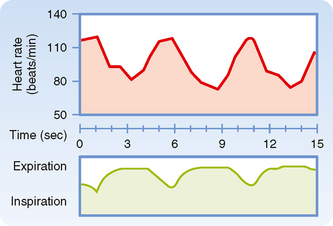
Recordings from cardiac autonomic nerves reveal that neural activity increases in the sympathetic fibers during inspiration and increases in the vagal fibers during expiration. The heart rate response to cessation of vagal stimulation is very quick because as already noted, the ACh released from the vagus nerves is rapidly hydrolyzed by cholinesterase. This short latency permits the heart rate to vary rhythmically at the respiratory frequency. Conversely, the norepinephrine released periodically at the sympathetic endings is removed very slowly. Therefore, the rhythmic variations in sympathetic activity that accompany inspiration do not induce any appreciable oscillatory changes in heart rate. Thus, respiratory sinus arrhythmia is almost entirely brought about by changes in vagal activity. In fact, respiratory sinus arrhythmia is exaggerated when vagal tone is enhanced.
Both reflex and central factors help initiate respiratory sinus arrhythmia (Fig. 18-8). Stretch receptors in the lungs are stimulated during inspiration, and this action leads to a reflex increase in heart rate. The afferent and efferent limbs of this reflex are located in the vagus nerves. Intrathoracic pressure also decreases during inspiration and thereby increases venous return to the right side of the heart (see Chapter 19). The consequent stretch of the right atrium elicits the Bainbridge reflex. After the time delay required for the increased venous return to reach the left side of the heart, left ventricular output increases and raises arterial blood pressure. This rise in blood pressure in turn reduces the heart rate through the baroreceptor reflex.
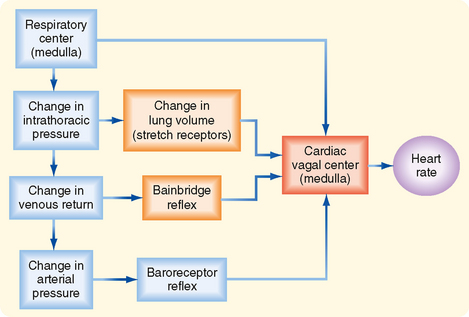
Central factors are also responsible for respiratory cardiac arrhythmia. The respiratory center in the medulla directly influences the cardiac autonomic centers (Fig. 18-8). In heart-lung bypass experiments, the chest is open, the lungs are collapsed, venous return is diverted to a pump-oxygenator, and arterial blood pressure is maintained at a constant level. In such experiments, rhythmic movement of the rib cage attests to the activity of the medullary respiratory centers. Such movement of the rib cage is often accompanied by rhythmic changes in heart rate at the respiratory frequency. This respiratory cardiac arrhythmia is almost certainly induced by a direct interaction between the respiratory and cardiac centers in the medulla.
Chemoreceptor Reflex
The cardiac response to peripheral chemoreceptor stimulation illustrates the complex interactions that may ensue when one stimulus excites two organ systems simultaneously. In intact animals, stimulation of the carotid chemoreceptors consistently increases ventilatory rate and depth (see Chapter 24), but ordinarily it changes the heart rate only slightly. The magnitude of the ventilatory response determines whether the heart rate increases or decreases as a result of carotid chemoreceptor stimulation. Mild chemoreceptor-induced stimulation of respiration decreases the heart rate moderately; more pronounced stimulation increases the heart rate only slightly. If the pulmonary response to chemoreceptor stimulation is blocked, the heart rate response may be greatly exaggerated, as described later.
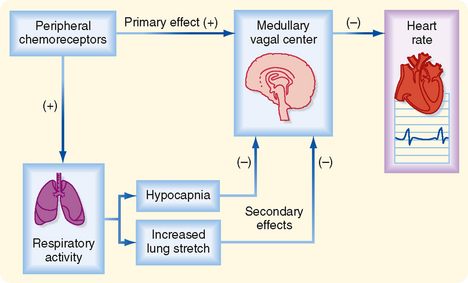
The cardiac response to peripheral chemoreceptor stimulation is the result of primary and secondary reflex mechanisms (Fig. 18-9). The principal effect of the primary reflex stimulation is to excite the medullary vagal center and thereby decrease the heart rate. The respiratory system mediates secondary reflex effects. The respiratory stimulation by arterial chemoreceptors tends to inhibit the medullary vagal center. This inhibition varies with the level of concomitant stimulation of respiration; small increases in respiration inhibit the vagal center slightly, whereas large increases in ventilation inhibit the vagal center more profoundly.
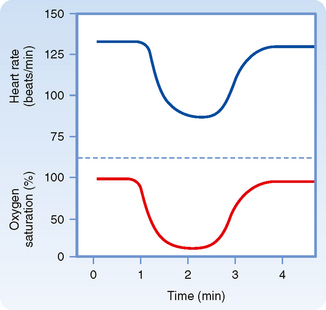
An example of the primary inhibitory influence is shown in Figure 18-10. In this example, the lungs are completely collapsed and blood oxygenation is accomplished with an artificial oxygenator. When the carotid chemoreceptors are stimulated, an intense bradycardia and some degree of AV block ensue. Such effects are mediated primarily by efferent vagal fibers.
The pulmonary hyperventilation that is ordinarily evoked by carotid chemoreceptor stimulation influences the heart rate secondarily, both by initiating more pronounced pulmonary inflation reflexes and by producing hypocapnia (Fig. 18-9). Both influences tend to depress the primary cardiac response to chemoreceptor stimulation and thereby accelerate the heart. Hence, when pulmonary hyperventilation is not prevented, the primary and secondary effects neutralize each other, and carotid chemoreceptor stimulation affects the heart rate only moderately.
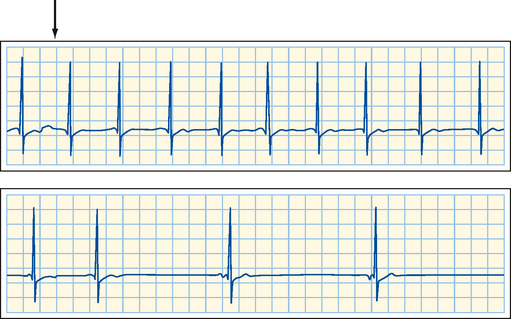
The electrocardiogram in Figure 18-11 was recorded from a quadriplegic patient who could not breathe spontaneously and required tracheal intubation and artificial respiration. When the tracheal catheter was briefly disconnected (near the beginning of the top strip in the figure) to permit nursing care, profound bradycardia quickly developed. The patient’s heart rate was 65 beats/min just before the tracheal catheter was disconnected. In less than 10 seconds after cessation of artificial respiration, his heart rate dropped to about 20 beats/min. This bradycardia could be prevented by blocking the effects of efferent vagal activity with atropine, and its onset could be delayed considerably by hyperventilating the patient before disconnecting the tracheal catheter.
REGULATION OF MYOCARDIAL PERFORMANCE
Intrinsic Regulation of Myocardial Performance
As noted previously, the heart can initiate its own beat in the absence of any nervous or hormonal control. The myocardium can also adapt to changing hemodynamic conditions by means of mechanisms that are intrinsic to cardiac muscle itself. For example, racing greyhounds with denervated hearts perform almost as well as those with intact innervation. Their maximal running speed decreases by only 5% after complete cardiac denervation. In these dogs, the threefold to fourfold increase in cardiac output during a race is achieved principally by an increase in stroke volume. Normally, the increase in cardiac output with exercise is accompanied by a proportionate increase in heart rate; stroke volume does not change much (see Chapter 19). This adaptation in the denervated heart is not achieved entirely by intrinsic mechanisms; circulating catecholamines undoubtedly contribute. For example, if β-adrenergic receptor antagonists are given to greyhounds with denervated hearts, their racing performance is severely impaired.
Ventricular receptors have been implicated in the initiation of vasovagal syncope, a feeling of lightheadedness or brief loss of consciousness that may be triggered by psychological or orthostatic stress. The ventricular receptors are believed to be stimulated by reduced ventricular filling volume combined with vigorous ventricular contraction. In a person standing quietly, ventricular filling is diminished because blood tends to pool in the veins in the abdomen and legs, as explained in Chapter 17. Consequently, the reduction in cardiac output and arterial blood pressure leads to a generalized increase in sympathetic neural activity via the baroreceptor reflex (Fig. 18-5). The enhanced sympathetic activity to the heart evokes a vigorous ventricular contraction that thereby stimulates the ventricular receptors. Excitation of the ventricular receptors is believed to initiate the autonomic neural changes that evoke vasovagal syncope, namely, a combination of a profound, vagally mediated bradycardia and generalized arteriolar vasodilation mediated by a reduction in sympathetic neural activity.
Frank-Starling Mechanism
About a century ago, the German physiologist Otto Frank and the English physiologist Ernest Starling independently studied the response of isolated hearts to changes in preload and afterload (see Chapter 16). When ventricular filling pressure (preload) is increased, ventricular volume increases progressively and after several beats attains a constant, larger volume. At equilibrium, the volume of blood ejected by the ventricles (stroke volume) with each heartbeat increases to equal the greater quantity of venous return to the right atrium.
The increased ventricular volume facilitates ventricular contraction and enables the ventricles to pump a greater stroke volume. This increase in ventricular volume is associated with an increase in length of the individual ventricular cardiac fibers. The increase in fiber length alters cardiac performance mainly by altering the number of myofilament cross-bridges that interact (see Chapter 16). More recent evidence indicates that the principal mechanism involves a stretch-induced change in the sensitivity of cardiac myofilaments to Ca++ (see Chapter 16). An optimal fiber length exists, however. Excessively high filling pressures that overstretch the myocardial fibers may depress rather than enhance the pumping capacity of the ventricles.
Starling also showed that isolated heart preparations could adapt to changes in the counterforce to the ventricular ejection of blood during systole (i.e., afterload). As the left ventricle contracts, it does not eject blood into the aorta until the ventricle has developed a pressure that just exceeds the prevailing aortic pressure (see Chapter 16). The aortic pressure during ventricular ejection essentially constitutes the left ventricular afterload. In Starling’s experiments, arterial pressure was controlled by a hydraulic device in the tubing that led from the ascending aorta to the right atrial blood reservoir. Venous return to the right atrium was held constant by maintaining the hydrostatic level of the blood reservoir. As Starling raised arterial pressure to a new, constant level, the left ventricle responded at first to the increased afterload by pumping a diminished stroke volume. Because venous return was held constant, the diminution in stroke volume was accompanied by a rise in ventricular diastolic volume, as well as by an increase in the length of the myocardial fibers. This change in end-diastolic fiber length finally enabled the ventricle to pump a normal stroke volume against the greater peripheral resistance. Again, a change in the number of cross-bridges between the thick and thin filaments probably contributes to this adaptation, but the major factor appears to be a stretch-induced change in the sensitivity of the contractile proteins to Ca++.
When cardiac compensation involves ventricular dilation, one must consider how the increased size of the ventricle affects the generation of intraventricular pressure. According to the Laplace relationship (see Chapter 17), if the ventricle enlarges, the force required by each myocardial fiber to generate a given intraventricular systolic pressure must be appreciably greater than that developed by the fibers in a ventricle of normal size. Thus, more energy is required for a dilated heart to perform a given amount of external work than for a normal-sized heart. Hence, computation of afterload on contracting myocardial fibers in the walls of the ventricles must consider ventricular dimensions along with intraventricular (and aortic) pressure.
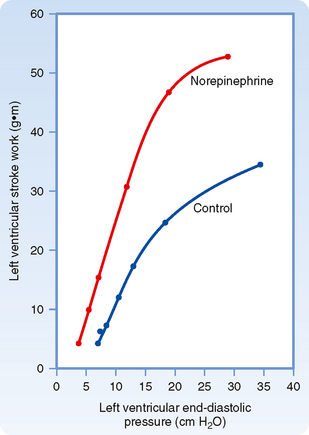
To assess changes in ventricular performance, the Frank-Starling mechanism is often represented by a family of ventricular function curves. To construct a control ventricular function curve, for example, blood volume is altered over a range of values, and stroke work (i.e., stroke volume × mean arterial pressure) and end-diastolic ventricular pressure are measured at each step. Similar observations are then made during the desired experimental intervention. For example, the ventricular function curve obtained during infusion of norepinephrine lies above and to the left of the control ventricular function curve (Fig. 18-12). Clearly, for a given level of left ventricular end-diastolic pressure (an index of preload), the left ventricle performs more work during the norepinephrine infusion than during control conditions. Hence, the upward and leftward shift of the ventricular function curve signifies improved ventricular contractility. Conversely, a shift downward and to the right indicates impaired contractility and a tendency toward cardiac failure.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


