- The quinolones are synthetic antibacterial agents which show greatest activity against Gram negative bacteria and are used in the treatment of urinary tract and respiratory infections.
- Resistance to quinolones arises due to alterations in the DNA gyrase (via mutations in the quinolone resistance-determining region (QRDR) of the gyrA gene), and similar mutations which decrease quinolone binding have been described in topoisomerase IV (in the parC gene).
- The quinolones are generally well tolerated, but some of the newer generations have been associated with serious adverse effects, such as hepatotoxicity and QT interval prolongation (cardiotoxicity).
- Some quinolones should not be routinely used in combination with theophylline because of the risk of developing theophylline toxicity.
- For further information, see Emmerson and Jones (2003) and Hooper (1998).
Antibacterial quinolones used clinically include nalidixic acid, ciprofloxacin, ofloxacin (racemic mixture of levofloxacin enantiomers), levofloxacin, norfloxacin, besifloxacin, and moxifloxacin; their structures are shown in Figure 2.1.1 and their therapeutic indications are listed in Table 2.1.1.
Figure 2.1.1 Quinolone antibacterials: nalidixic acid (first generation), ciprofloxacin and norfloxacin (second generation), levofloxacin (third generation), and besifloxacin and moxifloxacin (fourth generation)
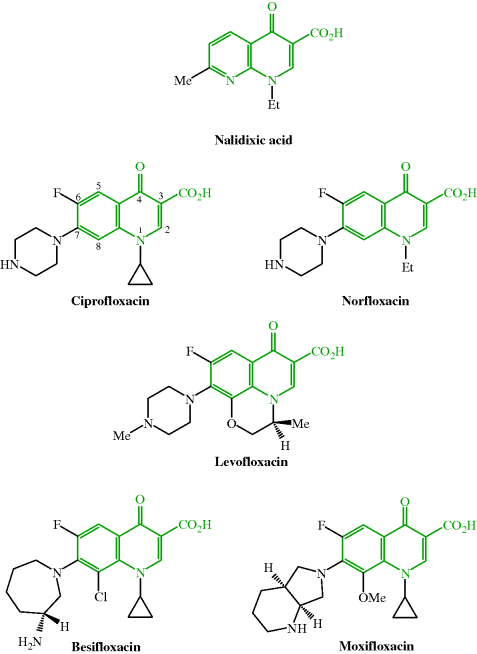
Table 2.1.1 Therapeutic indications for the quinolone antibacterials.
| Quinolone antibacterial | Indications |
| Nalidixic acid | Urinary tract infections (UTIs) |
| Ciprofloxacin | Respiratory tract infections (RTIs), UTIs, anthrax treatment, gonorrhoea, chronic prostatitis, prophylaxis of meningitis |
| Ofloxacin | UTIs, RTIs, gonorrhoea, urethritis |
| Levofloxacin | Community-acquired pneumonia |
| Norfloxacin | UTIs |
| Besifloxacin | Bacterial conjunctivitis |
| Moxifloxacin | Sinusitis, community-acquired pneumonia, exacerbations of chronic bronchitis |
2.1.1 Discovery
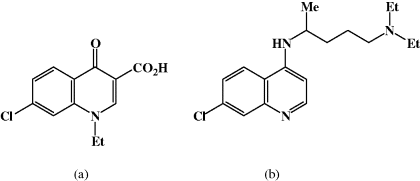
Nalidixic acid was discovered in 1962, as a result of a drug discovery project at Sterling Winthrop, which used as the lead compound a quinolone impurity (Figure 2.1.2a) isolated during the synthesis and purification of the antimalarial agent, chloroquine (Figure 2.1.2b). Nalidixic acid, like other first-generation quinolones, was found to have weak antibacterial (bactericidal) activity and is now only used for the treatment of urinary tract infections (UTIs) caused by Gram negative bacteria, as is norfloxacin, a second-generation quinolone.
Figure 2.1.2 (a) Quinolone impurity. (b) Chloroquine (Reprinted from J. M. Domagala, J. Antimicrob. Chemother., 33, 685–706, 1994, with permission of the Oxford University Press.)
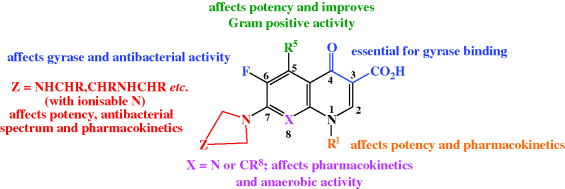
All second-generation and later quinolones contain, in addition to the 3-carboxyl-substituted quinolin-4-one essential for biological activity, a fluorine at the 6-position and a cyclic amine at the 7-position, both of which were quickly discovered to increase the antibacterial activity of these agents during structure–activity relationship (SAR) studies (Domagala, 1994). The nature of the cyclic amine, along with the introduction of other substituents, influences the antibacterial spectrum of activity (including activity against Gram positive organisms), as well as the physicochemical and pharmacokinetic properties of these synthetic antibacterials (Figure 2.1.3); five- and six-membered rings confer greater activity (although besifloxacin, of course, has a seven-membered (azepine) ring at this position), with a six-membered ring improving the activity against Gram negative species, for example, the piperazine ring in ciprofloxacin, and ring alkylation improving both the Gram positive activity and the half-life. (For more on the discovery of the quinolones, see Rádl, 1996.)
2.1.2 Synthesis
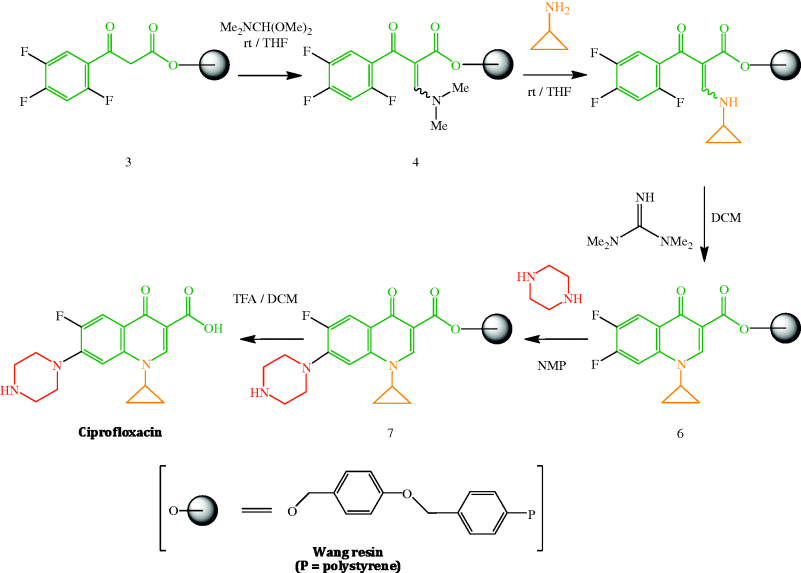
As many thousands of quinolones have been synthesised and tested, there are a large number of literature syntheses (including patents) of this class of antibacterial agent and it is not within the scope of this section to discuss their various merits. One particularly elegant synthesis, however, which builds upon earlier synthetic strategies but on a solid phase support, is the use of DIVERSOMER technology by MacDonald et al. (1996), which is illustrated in Scheme 2.1.1 for the preparation of ciprofloxacin. This methodology has a number of advantages and a range of fluoroquinolones can be prepared from common intermediates 4 (from which various N1-substituted derivatives can be obtained through variation of the amine used in the coupling step) and 6 (from which various cyclic amines can be introduced at position 7). Solid phase organic synthesis allows optimal yields to be obtained at each stage through the use of excess reagents, which are simply washed away after each step, with the intermediates remaining linked to the Wang resin until the final cleavage of the resin-bound fluoroquinolone 7 with trifluoroacetic acid. In addition to the introduction of the N1-substituent and the heterocycle (piperidine in this example) at position 7, other key steps are the reaction of the active methylene group in β-ketoester 3 with dimethylformamide dimethylacetal to introduce C-2 and an amine group, a transamination to the cyclopropylamine derivative 5, the base-catalysed cyclisation of the amine 5 via a nucleophilic aromatic substitution (which is facilitated by the presence of the electron-withdrawing fluorine atoms and the ortho-carbonyl group on the aryl ring, as well as the fluoro leaving group), and the final cleavage step (Scheme 2.1.1).
Scheme 2.1.1 Solid phase synthesis of ciprofloxacin (rt, room temperature; THF, tetrahydrofuran; DCM, dichloromethane; NMP, N-methylpyrrolidone; TFA, trifluoroacetic acid) (MacDonald et al., 1996)
As can be seen from Figure 2.1.1, the heterocyclic ring attached to position 7 of the quinolone nucleus increases in complexity in the structures of the third- and fourth-generation fluoroquinolones, with many of these later derivatives having a number of stereogenic centres. Ofloxacin is a mixture of enantiomers, but each of these optical isomers will have been synthesised and tested separately as part of the licensing application to the relevant agency – the European Medicines Agency (EMA), the Medicines and Healthcare products Regulatory Agency (MHRA), the Food and Drug Administration (FDA), the Australian Therapeutic Goods Administration (TGA), and so on. In general, the greater the number of stereogenic centres in a drug candidate, the more complex the synthesis, with each stereoisomer having to be prepared, isolated, characterised, and tested. As chiral synthesis is generally expensive (unless a route which utilises a relatively cheap and easily available chiral natural product can be employed), this all adds to the cost of the drug discovery process, and such an increase in the complexity of these heterocyclic side chains must be justified by an increase in the efficacy or antibacterial spectrum of the agent, or a decrease in their side effects.
2.1.3 Bioavailability
There have been many investigations into the bioavailability of the quinolones; much of this work has focussed upon ciprofloxacin, but others are currently under scrutiny as the newer-generation quinolones become more popular.
If you reinspect the general structure of an antibacterial quinolone (Figure 2.1.3) for ionisable groups that could affect the bioavailability, you will note the carboxylic acid at position 3, which is essential (as the negatively charged carboxylate) for binding to the target, and the ionisable amine group on position 7. Together, these groups confer a zwitterionic nature that leads to the quinolones being largely ionised in all physiological compartments. Despite the predominance of ionised species in the gastrointestinal (GI) tract, the quinolones are well absorbed after oral administration, with good overall bioavailability, typically 70% (e.g. ciprofloxacin) to 99% (e.g. levofloxacin) (Blondeau, 1999; Wright et al., 2000; Rodvold and Neuhauser, 2001), with measured logPapp values (octanol/buffer at pH 7.0) in the range of approximately −1.6 (norfloxacin, ciprofloxacin) to 2.5 (nalidixic acid) (Ross et al., 1992; Bermejo, 1999; Piddock et al., 2001; Perez et al., 2002). While the majority of quinolones have negative apparent partition coefficients, the third- and fourth-generation quinolones are reported to be significantly more lipophilic and better absorbed, after oral, ophthalmic, or respiratory system delivery, presumably due to the presence in their structures of more lipophilic groups (Perletti et al., 2009).
Absorption of the quinolones is known to be reduced in the presence of divalent or trivalent metal cations, such as Mg2+, Ca2+, Al3+, or Fe3+, due to chelation of the cation to the quinolone carbonyl and carboxylate groups; the bioavailability of the quinolones is, therefore, limited by co-administration with metal cation-rich products, such as antacid preparations and iron supplements (more on this later, in Subsection 2.1.8).
For these concentration-dependent antibiotics, the higher the serum concentration, the greater the efficacy of bactericidal action. The most important parameters for these agents are the serum peak concentration (Cmax) and the area under the serum concentration–time curve (AUC) when compared to the minimum inhibitory concentration (MIC), alternatively expressed as the maximum concentration achieved and maintained over a specific time when compared to the concentration required for bactericidal effect. The half-life of the quinolone (t½) impacts upon both of these parameters, as it represents the length of time the quinolone can be expected to exert its bactericidal effect.
The early quinolones, such as ciprofloxacin and norfloxacin, have relatively short half-lives (3–5 hours), while the third- and fourth-generation quinolones have extended serum half-lives (e.g. 7.4 hours for levofloxacin and 9.6 hours for moxifloxacin), which enables single daily dosing. The Cmax values for quinolones vary from 1.1 to 8.7 mg/L and the AUC values lie in the range 10–75 mg/h/L. The quinolones show exquisite potency towards a wide range of Gram negative bacteria, with MICs in the ng/mL range, and reasonable activity against many Gram positive bacteria (MICs in the mg/mL range), although some of the newer quinolones, such as moxifloxacin, are significantly more potent against this group of bacteria. The excellent MICs and good Cmax and AUC values lead to strong pharmacokinetic profiles for the quinolones overall and the newer quinolones tend to have enhanced parameters for improved delivery and efficacy (Blondeau, 1999; Wright et al., 2000; Cheng et al., 2007).
Quinolone distribution is influenced by protein binding, typically in the range of 20–70%. The volume of distribution (Vd) varies in the range 1.1–5.1 L/kg, newer quinolones usually having higher values and better tissue penetration, enabling their use across a wider range of infections. The quinolones are subject to comparatively little metabolism, most being excreted largely unchanged in the urine, although moxifloxacin exhibits significant faecal excretion (~10%) (Blondeau, 1999; Wright et al., 2000).
The high urinary excretion of most quinolones, along with their short half-lives, highlights the treatment of UTIs as a possible indication. The third- and fourth-generation fluoroquinolones show greater versatility for the treatment of systemic infections, due to their superior activity across a wider range of Gram positive bacteria and their improved Vd and uptake into many cells, due to their increased lipophilicity, enabling, for example, the treatment of prostatic infections (Perletti et al., 2009).
2.1.3.1 Prodrugs
As the bioavailability of the quinolones is generally very good, there has been little need for quinolone prodrugs, with the only one to have been licensed and used clinically being alatrofloxacin, a dialanyl-derivative of trovafloxacin, a broad-spectrum quinolone with excellent activity against a wide range of both Gram negative and Gram positive bacteria, including MRSA, but whose low aqueous solubility has limited its IV use. The two L-alanine amino acid residues confer considerable aqueous solubility to alatrofloxacin, thus allowing parenteral delivery of the prodrug, which undergoes rapid systemic hydrolysis to the parent, trovafloxacin (Scheme 2.1.2) (Melnik et al., 1998; Vaudaux et al., 2002).
Scheme 2.1.2 Structure of the prodrug alatrofloxacin and its hydrolysis to its parent, trovafloxacin
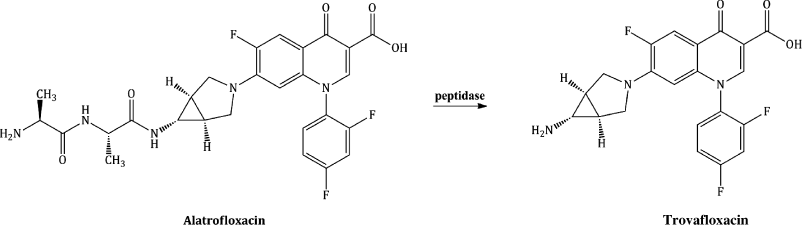
Trovafloxacin and the prodrug alatrofloxacin were withdrawn from clinical use in Europe in 1999 (but are still available in the USA) due to unpredictable and severe hepatotoxicity, with a high risk of fatality (Pannu et al., 2001).
Two successful phase III clinical trials of the prodrug prulifloxacin (Figure 2.1.4), designed for oral administration, were recently completed. Prulifloxacin delivers the parent drug, ulifloxacin, immediately and quantitatively after efficient absorption. This thiazeto-quinolone antibacterial agent has potent activity against a wide range of both Gram negative and Gram positive bacteria, with MICs in the ng/mL range.
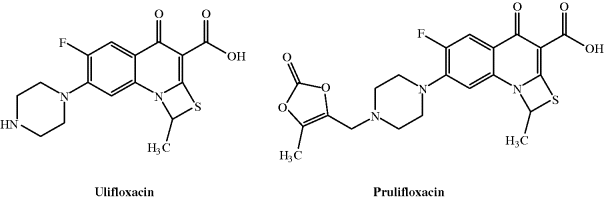
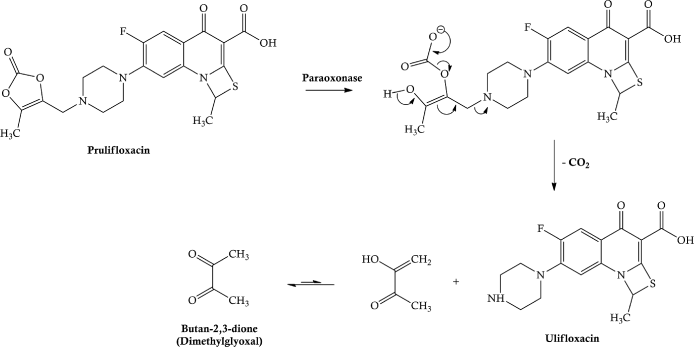
Prulifloxacin is well-absorbed in the upper small intestine and metabolised to the parent, ulifloxacin, so rapidly that the prodrug is not detected in the systemic circulation. Interestingly, ulifloxacin accumulates in the liver and kidney, with a relatively high concentration in other organs, such as the spleen, pancreas, and lungs, yet a low distribution into the central nervous system (CNS) (Matera, 2006). The enzyme paraoxonase has a major role in the hydrolysis of this unusual prodrug (Tougou et al., 1998), the postulated mechanism for which is shown in Scheme 2.1.3.
2.1.3.2 Bacterial Uptake of Quinolones
As we will see in the next subsection, the target for quinolones is found in the cytoplasm, so these agents must cross the bacterial cell wall and enter the cell to exert their antibacterial activity. They readily cross the cell wall of Gram positive bacteria, such as staphyloccocci, by passive diffusion and even though efflux mechanisms for hydrophilic quinolones in Gram positive bacteria have been identified, they are well accumulated rapidly into these bacteria, to a higher concentration than is achieved in Gram negative bacteria. The more complex cell wall of Gram negative bacteria is not amenable to passive diffusion and quinolones are taken into these bacteria through the outer membrance porins, facilitated for the more lipophilic examples by diffusion through the outer membrane. The physicochemical properties of each antibacterial agent, such as lipophilicity and molecular mass, have been correlated to the kinetics of uptake and, for some species, the antibacterial effect upon Gram negative bacteria (Piddock et al., 2001).
2.1.4 Mode of Action and Selectivity
As discussed in the introductory section, if we know the mode of action of a class of antibacterial agents then we will be able to understand how their selectivity arises (i.e. what cellular process the agents target in prokaryotic cells and how this differs from eukaryotic cells) and how bacteria will develop resistance to them.
As you will have gathered from the title of this section, the quinolones exert their antibacterial activity by targeting DNA synthesis, so in order to understand how they do this, we need to look at the processes involved in DNA replication. We will also need to focus on how the quinolones can do this selectively in prokaryotic (bacterial) cells as, of course, this process will also be taking place in eukaryotic (mammalian) cells.
As we saw in Subsection 1.1.3, DNA replication is a complex process during which the two strands of the double helix separate and each strand acts as a template for the synthesis of complementary DNA strands. This process occurs at multiple, specific locations (origins) along the DNA strand, with each region of new DNA synthesis involving many proteins, which catalyse the individual steps involved in this process – the separation of the two strands at the origin to give a replication fork (DNA helicase); the synthesis and binding of a short primer DNA strand (DNA primase); DNA synthesis (DNA polymerase); and, finally, the proofreading and error-checking process to ensure the new DNA strand’s fidelity – that is, that this strand (red) is exactly complementary to the template (black) strand (DNA polymerase and endonucleases) (Figure 1.1.4, Subsection 1.1.3). As you can see from Figure 1.1.4, the replication fork is a busy place, but for the purposes of the discussion of the mode of action of the quinolones, we can concentrate on the role of two of the enzymes shown – helicase and topoisomerase (Duguet, 1997).
DNA helicase is the enzyme which separates the DNA strands and in doing this, as a result of the right-handed helical nature of DNA, produces positive supercoils (knots) ahead of the replication site. In order for DNA replication to proceed, these supercoils must be removed by topoisomerases, relaxing the chain. By catalysing the formation of negative supercoils, through the cutting of the DNA chain(s) and the passing of one strand through the other, these enzymes remove the positive supercoils and give a tension-free DNA double helix, so that the replication process can continue. Type I topoisomerases relax DNA by cutting one of the DNA strands, while, you’ve guessed it, type II cut both strands (Champoux, 2001). For the purpose of understanding the mode of action of the quinolones, it is the type II topoisomerases (DNA gyrase in Gram negative and topoisomerase IV in Gram positive bacteria) that are important (Ruiz, 2003). By binding to these enzymes, the quinolones interfere with bacterial DNA replication, and so with cell division (Figure 2.1.5). So far so good, but two questions you have probably already asked yourself are: given that mammalian cells must also relax supercoiled DNA, why are the quinolones selective antibacterial agents, and, if the quinolones affect cell division (i.e. the division of mature cells), why are they bactericidal rather than bacteriostatic? Good questions! First, mammalian cells do not have DNA gyrase or topoisomerase IV (they do have topoisomerases I and II, but quinolones do not bind to these enzymes), hence these agents have some selectivity as inhibitors of prokaryotic DNA replication. The fact that these agents are bactericidal rather than bacteriostatic is more difficult to address and is unlikely to be due to the inhibition of DNA synthesis, as the concentration of a quinolone required to block DNA synthesis is much lower than that required to kill cells.
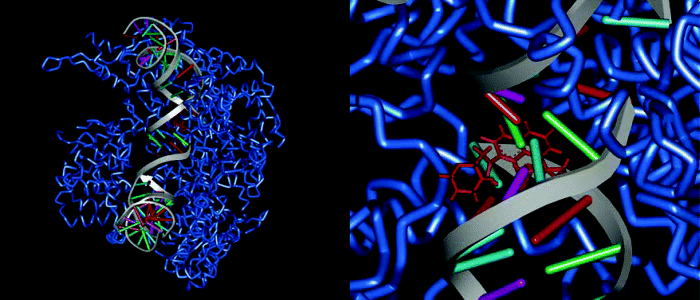
Figure 2.1.5 A DNA-topoisomerase IV (blue) complex from Streptococcus pneumoniae stabilized by moxifloxacin (red); PDB code 3FOF (Laponogov et al., 2009)
One of the possible explanations for the bactericidal effect of quinolones is the generation of DNA ends (effectively DNA double-strand breaks) through the formation of reversible ternary ‘cleavable’ complexes (quinolone-topoisomerase-DNA) and the resultant release of the DNA ends from the constraints of the topoisomerase (Figure 2.1.6) (Froehlich-Ammon and Osheroff, 1995). Such double-strand DNA breaks, when generated by other means, are known to be lethal to cells (Drlica and Zhao, 1997).
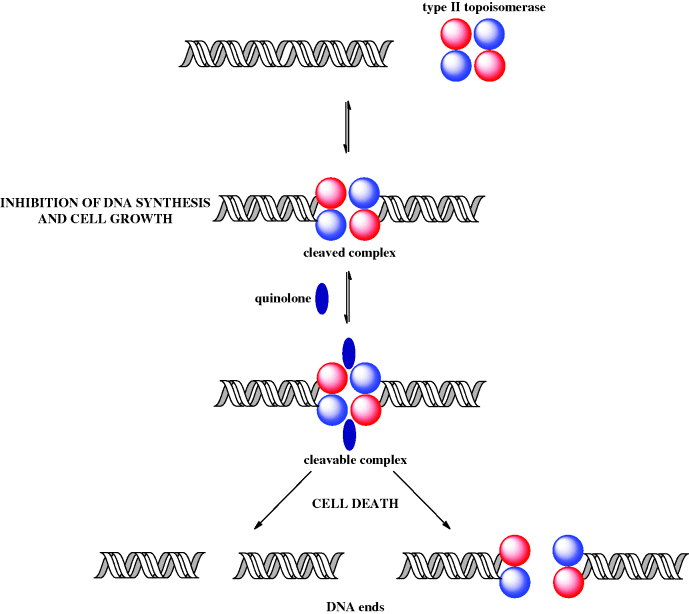
2.1.5 Bacterial Resistance
As we suggested previously, knowing the mode of action of an antibacterial agent will help us to understand the way in which bacteria develop resistance, as the specific resistance mechanisms will be related to interference with the processes targeted by the antibacterial agent. As described in the previous subsection, the cellular targets of the quinolones are the bacterial DNA gyrase and topoisomerase IV, both of which are heterotetrameric in structure; that is, formed from four subunits: A2B2 for DNA gyrase, C2E2 for topoisomerase IV. (The structure of these enzymes represents a key difference from the mammalian type I (monomer) and II (homodimer) topoisomerases and this may account for the binding of the quinolones to the prokaryotic, but not the eukaryotic, enzymes.) As might be expected, bacteria have developed resistance to the quinolones through alterations in the target enzymes – alterations to the DNA gyrase occur via mutations in the quinolone resistance-determining region (QRDR) of the gyrA gene, which encodes the two A subunits of the tetrameric enzyme (gyrB encodes the two B subunits), while similar mutations which decrease quinolone binding have been described in topoisomerase IV (in the parC gene, which codes for the two C subunits (parE codes for the two E subunits)) (Ruiz, 2003).
Both Pseudomonas aeruginosa (Gram negative) (Nikaido, 1996) and Staphylococcus aureus (Gram positive) exhibit well-characterised efflux pumps for the quinolones (Putman et al., 2000), but bacteria have also developed resistance to the quinolones through decreased cellular uptake due to the impermeability of the cell membrane. Quinolones cross the outer membrane via specific porins (all quinolones) or by diffusion through the phospholipid bilayer (hydrophobic quinolones only). Porins are protein channels which allow passive diffusion of a specific agent across the cell membrane; for example, E. coli bacteria have three main porins, a decrease in the level of one of which (OmpF) is associated with an increase in resistance to the quinolones (Cohen et al., 1989). In addition, the outer membrane of P. aeruginosa has very low permeability to small hydrophobic molecules, giving this bacterium some intrinsic resistance to the quinolones (Yoshimura and Nikaido, 1982).
2.1.6 Clinical Applications
2.1.6.1 Spectrum of Activity
The quinolones currently available generally have greatest activity against Gram negative bacteria, with the most susceptible organisms including members of Enterobacteriaceae, Neisseria species and Haemophilus species (Hooper, 1998); Pseudomonas aeruginosa and Acinetobacter species are also susceptible.
Activity against Gram positive bacteria has, over the years, been debatable, but the newer-generation quinolones, such as levofloxacin and moxifloxacin, appear to show improved activity; for example, moxifloxacin shows good activity against Staphylococcus aureus (meticillin-susceptible) and Streptococccus pneumoniae (which are Gram positive bacteria).
2.1.6.2 Urinary Tract Infections (UTIs)
As mentioned previously, quinolones show good activity against Gram negative bacteria, with, for example, ciprofloxacin particularly active against Salmonella, Shigella, Campylobacter, Neisseria, and Pseudomonas species. This is important because UTIs, which are relatively common, especially in women, are frequently caused by Gram negative bacteria. It is estimated that 40–50% of women will experience a UTI in their lifetime; the infection is much less common in men, as bacteria can pass up the shorter urethra of a woman much more readily than that of a man. There is also evidence suggesting that prostatic fluid, present in men, has antibacterial properties and provides an additional defence mechanism against UTIs.
Generally, the first-line treatment for uncomplicated UTIs is either trimethoprim (a DHFR inhibitor; see Section 3.2) or nitrofurantoin (which is similar in mode of action to metronidazole; see Section 2.3) for three to seven days (three days treatment is usually sufficient), but if the first-line therapy fails, or bacterial culture suggests there is resistance to these agents, a quinolone can be used. Studies show that ciprofloxacin is highly active against the most common pathogens causing UTIs (e.g. E. coli) (Farrell et al., 2003) and this activity appears to be evident for all of the quinolones, as a Cochrane review found no significant differences in clinical or microbiological activity between quinolones for the treatment of uncomplicated acute cystitis (Rafalsky et al., 2006). UTIs in men, as discussed previously, are less common and generally occur as a result of an anatomical or functional problem with the genitourinary tract, so are considered more serious. UTIs in men are therefore often considered to be ‘complicated’ (as opposed to the uncomplicated UTIs that frequently occur in women) and may be treated with a two-week course of a quinolone (SIGN, 2006).
Quinolones from all generations, including nalidixic acid, ciprofloxacin, ofloxacin, levofloxacin and norfloxacin, are used in the management of UTIs. In elderly patients, when compliance is often a problem, a quinolone with a once-daily dosing regimen, such as ofloxacin, would be preferred over one with a twice-daily dosing regimen, such as ciprofloxacin. Moxifloxacin, a fourth-generation quinolone, is not used in the routine management of uncomplicated UTIs, because it has been associated with the risk of life-threatening hepatotoxicity (Verma et al., 2009). As a result of this potentially serious adverse effect, moxifloxacin is reserved for the treatment of sinusitis, community-acquired pneumonia, or cases of chronic bronchitis that have failed to respond to other antibacterial therapies.
In addition to UTIs, quinolones can be used to treat bacterial prostatitis (inflammation of the prostate gland), which can present as either an acute or a chronic condition, with bacterial infection accounting for the majority of acute cases. Symptoms often include fever (a general sign of infection), dysuria (painful urination), low abdominal pain, pain on ejaculation, and urethral discharge. Ciprofloxacin (and other quinolones) are often preferred to other antibacterial agents for the treatment of bacterial prostatitis, because they penetrate well into the prostate tissue, a property few other antibacterial agents possess (Wolfson and Hooper, 1989). In the case of chronic bacterial prostatitis, extended courses of quinolones (in some cases up to six weeks) are often required.
2.1.6.3 Respiratory Tract Infections (RTIs)
Quinolones are also used clinically in the treatment of respiratory tract infections (RTIs). These infections, frequently acquired in the community, are often caused by pathogens such as S. pneumoniae (Gram positive) and H. influenzae (Gram negative). Ciprofloxacin and ofloxacin show good activity against the Gram negative H. influenzae, but reduced activity against the Gram positive S. pneumoniae, and these agents should therefore not be used in the treatment of pneumococcal pneumonia. The newer agents, such as levofloxacin (third generation) and moxifloxacin (fourth generation), show improved activity against S. pneumoniae and thus provide a useful therapeutic alternative to the earlier quinolones. A recent meta-analysis, concerning the treatment of exacerbations of chronic bronchitis, concluded that the clinical success rate was higher with moxifloxacin than for the standard antibiotic therapy (such as amoxicillin or clarithromycin) (Miravitlles et al., 2007).
At present, quinolones are not the recommended first-line treatment for the management of uncomplicated community-acquired pneumonia, which is often managed successfully with amoxicillin (or erythromycin, if the patient is allergic to penicillin; see Section 4.2). In these cases, quinolones are not the recommended first-line treatment, in order to minimise the threat of bacterial resistance, allowing them to be kept in reserve for more serious infections.
In the case of hospital-acquired pneumonia, a quinolone such as ciprofloxacin, ofloxacin, levofloxacin, or moxifloxacin may be used. Hospital-acquired pneumonia can be defined as pneumonia which occurs 48 hours after admission to hospital and is often caused by S. aureus (including meticillin-resistant Staphylococcus aureus (MRSA)) or, in some cases, opportunistic Gram negative bacilli, such as P. aeruginosa. Unfortunately, as outlined previously, some strains of P. aeruginosa have developed resistance to the quinolones through the expression of efflux pumps, which limits their role in the treatment of some hospital-acquired pneumonias. Ultimately, the choice of agent for the treatment of hospital-acquired pneumonia is dependent on both bacterial sensitivity and the local antibiotic policy of the hospital (we will discuss this more in Subsection 5.1.6 when we outline the clinical uses of the β-lactams).
2.1.6.4 Sexually Transmitted Infections (STIs)
Some quinolones also have good activity against strains of Neisseria gonorrhoeae (the organism responsible for causing the sexually transmitted infection gonorrhoea). N. gonorrhoeae is a Gram negative organism which is found in the semen of infected men and the vaginal fluids of infected women. Around half of women infected with gonorrhoea can be asymptomatic, while men are much more likely to present with symptoms (British Association for Sexual Health and HIV, 2005).
One-off, single-dose treatments (as a tablet) of ciprofloxacin or ofloxacin are preferred in these instances to assure compliance with therapy, which is advantageous as it will help reduce the risk of further sexual transmission of the infection. There have also been reports suggesting that strains of N. gonorrhoeae are now beginning to show signs of resistance to ciprofloxacin and ofloxacin (Abeyewickereme et al., 1996); if this is confirmed then cefixime, a third-generation cephalosporin, can be used as a single (oral) dose to treat the infection, although this is an unlicensed indication for cefixime.
2.1.6.5 Tuberculosis
Tuberculosis is caused by Mycobacterium tuberculosis and is a major public health concern, particularly in developing countries, where multidrug- (MDR-TB) and extensively drug-resistant (XDR-TB) strains have emerged. If left untreated or not treated appropriately, tuberculosis can be fatal, and it is claimed that tuberculosis causes more adult deaths worldwide than any other infection (Kochi, 1991). Quinolones have activity against M. tuberculosis, but until recently it has not been clear what role they have in the treatment of tuberculosis.
A recent, evidence-based Cochrane review concluded that the routine addition or substitution of quinolones to conventional antituberculosis chemotherapy is not recommended (Ziganshina and Squire, 2008). In addition, when ciprofloxacin was used to treat drug-sensitive tuberculosis, it was found to actually increase the relapse rate of infection when substituted into first-line antituberculosis chemotherapy (Ziganshina and Squire, 2008).
As with all infectious diseases, resistance to conventional antimicrobial therapy is becoming a significant clinical problem, as illustrated by the rise of multidrug-resistant tuberculosis (MDR-TB). When a strain of M. tuberculosis develops resistance to rifampicin and isoniazid, it is said to be multiple drug-resistant. MDR-TB is very costly and difficult to manage, even in countries with highly developed health care systems, and threatens to jeopardise overall tuberculosis control efforts (Mukherjee et al., 2004). Quinolones appear to have a role in the management of MDR-TB, with the newer generations, such as moxifloxacin, showing increased in vitro activity compared to the older generations. Indeed, the quinolones have been included in MDR-TB drug regimens since the late 1980s and their unique mode of action, through DNA-gyrase/topoisomerase inhibition, would appear to offer a welcome additional therapeutic target to those of the drugs found in regular regimens. The exact role of quinolones in the management of TB remains controversial, however; for example, one small randomised controlled trial compared the standard antituberculosis drug regimens to the standard antituberculosis regimens plus levofloxacin in the management of TB (the trial included drug-resistant and drug-susceptible patients) (el-Sadr et al., 1998) and found that there was no statistically significant difference in clinical outcomes between the two regimens. The recent Cochrane review, however, acknowledges that the quinolones have a role in the management of MDR-TB, but suggests that further trials, particularly involving studies on the newer-generation quinolones (such as moxifloxacin), are needed to establish exactly what that role is (Ziganshina and Squire, 2008).
2.1.6.6 Anthrax
Ciprofloxacin has also had an important role to play in the treatment of anthrax, which was used as a biological weapon in a relatively recent terrorist attack. This occurred just after the September 11th terrorist attacks in 2001; anthrax spores were sent through the US postal service to several media outlets and two US senators. As a result of this bioterrorist attack, at least five people died, and the number of victims (who required antibiotics) was as many as 68 people (Cymet and Kerkvliet, 2004). Anthrax, caused by Bacillus anthracis, is often fatal if left untreated. Ciprofloxacin shows activity against B. anthracis and, as a result, the FDA recommends the use of ciprofloxacin to treat anyone who has been exposed to anthrax spores (Doganay and Aydin, 1991), with a recommended dose of ciprofloxacin of 500 mg twice daily for 60 days (alternative agents are penicillins and doxycycline, but the drug of choice remains ciprofloxacin). The treatment period of 60 days remains a problem in terms of patient compliance and studies have demonstrated that eradication of the spore burden can be achieved after 35 days of treatment (95% of patients). Spore burden eradication was achieved in 99.5% of patients after 110 days’ treatment, so 60 days remains a suitable compromise (Drusano et al., 2008). Safety data are limited for the use of ciprofloxacin during pregnancy and lactation, and as result the US Centers for Disease Control and Prevention (CDC) recommended amoxicillin in pregnant and lactating mothers, when the strain is penicillin-sensitive. Retrospective safety data show that exposure of the developing foetus to norfloxacin, ciprofloxacin, and ofloxacin is not associated with an increased risk of malformations: 2.2% rate of malformations in the quinolone group compared to 2.6% in the control group (Loebstein et al., 1998), although the rate of spontaneous abortions was higher in the quinolone-treated group of women, with a relative risk of 4.50. As data are still limited, the agent of choice in pregnant women remains penicillin.
2.1.6.7 Miscellaneous
Ciprofloxacin is often used to treat diarrhoea, which is one of the most common illnesses affecting travellers, particularly in developing countries. Travellers’ diarrhoea is often primarily infective in origin, with E. coli, Salmonella, and Campylobacter species all implicated. A single 500 mg dose of ciprofloxacin (or levofloxacin) is sufficient to reduce the severity of symptoms and also the duration of diarrhoea (Salam et al., 1994; Sanders et al., 2007). While travellers’ diarrhoea is usually self-limiting, such single-dose quinolone therapy helps to reduce the risk of developing complications, such as dehydration, which is particularly common in older people.
Quinolones are also used for the treatment of enteric fever (also known as typhoid or paratyphoid fever) caused by Salmonella typhi and Salmonella paratyphi. Quinolones are indicated for treating drug-sensitive and multiple drug-resistant (when there is resistance to chloramphenicol, amoxicillin, and co-trimoxazole) S. typhi and S. paratyphi in children and adults. Enteric fever is a major public health concern and has reached endemic status in many areas of the developing world. A recent meta-analysis published in the British Medical Journal and a Cochrane review concluded that quinolones may be better in helping to prevent clinical relapse of enteric fever than chloramphenicol, although data for recommending quinolones as first-line treatment in children are limited (Thaver et al., 2008, 2009).
There have also been developments in expanding the therapeutic use of the quinolones already on the market. A good example of this is the recent development of ciprofloxacin eye drops and eye ointment and ofloxacin eye drops, marketed in the UK as Ciloxin and Oftaquix, respectively. Ciloxin eye drops are licensed for the treatment of corneal ulcers and superficial eye infections, while Ciloxin eye ointment is licensed to treat corneal ulcers, conjunctivitis, and blepharitis. These products are effective against P. aeruginosa, an organism that can cause corneal ulceration, which, if left untreated, can lead to blindness. Oftaquix is licensed for the topical treatment of bacterial external ocular infections caused by microorganisms susceptible to levofloxacin. All of these products are not recommended for use in children under one year of age, as the clinical experience with such products is limited.
In addition, quinolone eye drops are also used to treat chronic otitis media (an inner-ear infection). This use may seem surprising, but ciprofloxacin or ofloxacin ear drops are only available on a named-patient basis (and must be ordered from a specialised company and so are difficult to obtain), so the eye formulations are often used as an unlicensed alternative. Ciprofloxacin and ofloxacin ear drops (or eye drops if the ear drops cannot be obtained) also offer an alternative to aminoglycoside antibiotic ear drops in the management of chronic otitis media in patients who have a perforated tympanic membrane.
2.1.7 Adverse Drug Reactions
Quinolones are generally well tolerated but mild side effects such as nausea, vomiting, dyspepsia, abdominal pain, diarrhoea, headache, and rashes have been reported. Less frequent side effects include blood disorders, such as eosinophilia (abnormally high numbers of eosinophils), leucopenia (decreased total number of white blood cells), and thrombocytopenia (decreased number of platelets), as well as anorexia, sleep disturbances, confusion, anxiety, depression, and hallucinations.
Very rare side effects (those that occur in less than 1 in 10 000 patients) include tendon inflammation and damage (mainly affecting the Achilles tendon), psychoses, and convulsions. With regard to tendon damage and inflammation, the Committee on Safety of Medicines (CSM) has advised that quinolones are contraindicated in patients with a history of tendon disorders associated with previous quinolone use. These adverse effects are rare, but are more common in elderly patients and in patients who have been treated with corticosteroids (Khaliq and Zhanel, 2005). Tendon damage is most common in the lower extremities, but has also been reported in the upper extremities. If a patient experiences any joint pain or inflammation, the quinolone should be discontinued immediately and care should be taken not to put excess weight on the affected extremity.
Quinolones should not be routinely used in children because of the hypothetical risk of arthropathy in weight-bearing joints. Although this risk has been established in immature animals but not, as yet, in humans, it is prudent to avoid the use of quinolones in children, unless the therapeutic benefit outweighs the potential risks associated with treatment. There are, however, some cases where the short-term use of a quinolone is warranted in children; for example, ciprofloxacin is often used to treat pseudomonal infections in cystic fibrosis patients over the age of five.
The side effects outlined above are all general to the quinolone family, but some side effects are specific to individual quinolones. For example, moxifloxacin (third generation) has been associated with life-threatening hepatotoxicity, resulting in fatalities (Verma et al., 2009). This adverse effect is considered to be very rare, but because of the serious consequences, patients are advised to contact their doctor if signs of hepatotoxicity, such as developing asthenia (physical weakness) associated with jaundice, dark urine, a bleeding tendency, or hepatic encephalopathy develop (Avelox summary of product characteristics).
2.1.7.1 Effect on QTc Wave and Proarrhythmic Potential
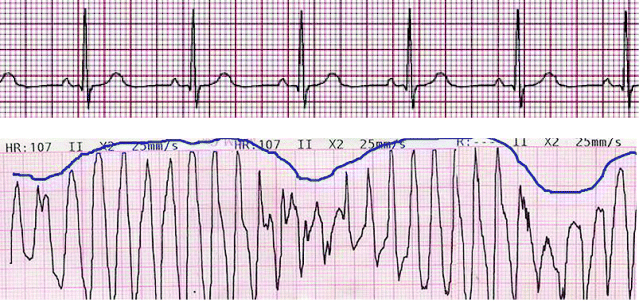
Some of the quinolones in clinical use demonstrate the adverse effect of prolongation of the QTc wave during administration, which can lead to torsade de pointes, a cardiac arrhythmia which can be life-threatening (Figure 2.1.7).
Figure 2.1.7 Traces showing a normal electrocardiogram (above) and torsade de pointes (below). The blue line shows the characteristic ‘twisting points’ associated with torsade de pointes (Reproduced from ‘Torsades de pointe’, Wikipedia, Online, [http://fr.academic.ru/dic.nsf/frwiki/1645373].)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


