Several amino acids have their metabolic pathways linked to the metabolism of other amino acids. These codependencies that link the pathways of amino acids become important when nutrient intake is limited or when metabolic requirements are increased. Two aspects of metabolism are reviewed here: (a) synthesis of amino acids and (b) amino acid degradation. Degradation serves two useful purposes: (a) production of energy from the oxidation of individual amino acids (≈4 kcal/g protein, almost the same energy production as for carbohydrate) and (b) conversion of amino acids into other products. The latter is also related to amino acid synthesis: the degradation pathway of one amino acid may be the synthetic pathway of another amino acid. The other important aspect of amino acid degradation is production of other nonamino acid, N-containing compounds in the body. The need for synthesis of these compounds may also drain the pools of their amino acid precursors and thus increase the need for these amino acids in the diet. When amino acids are degraded for energy rather than converted to other compounds, the ultimate products become CO2, water, and urea. The CO2 and water are produced through classical pathways of intermediary metabolism involving the tricarboxylic acid (TCA) cycle. The urea is produced because other forms of waste N, such as ammonia (NH3), are toxic if their levels rise in the blood and inside cells. For mammals, urea production is a means of removal of waste N from the oxidation of amino acids in the form of a nontoxic, water-soluble compound.
More detailed descriptions of the amino acid pathways can be found in standard textbooks of biochemistry. Keep in mind when consulting such texts that pathways for nonmammalian systems (e.g., Escherichia coli and yeast) are often presented, and these pathways often have little importance to human biochemistry. When consulting reference material, the reader needs to be aware of the system of life from which the metabolic pathways and enzymes are being discussed. The discussion here is relevant to human biochemistry. Presented first is a discussion of the routes of degradation of each amino acid when the pathway is directed toward oxidation of the amino acid for energy. Next follows a discussion of pathways of amino acid synthesis, and finally the use of amino acids for other important compounds in the body is described.
Amino Acid Degradation Pathways
Complete amino acid degradation ends up with the production of N, which is removed by incorporation into urea. Carbon skeletons are eventually oxidized as CO
2 via the TCA cycle (also known as the Krebs cycle or the citric acid cycle). The inputs to the cycle are acetyl-coenzyme A (CoA) and oxaloacetate forming citrate, which is degraded to α-ketoglutarate and then to oxaloacetate. The carbon skeletons from amino acid may enter the Krebs cycle via acetate as acetyl-CoA or via oxaloacetate/α-ketoglutarate. These latter two precursors are direct metabolites of the amino acids aspartate and glutamate. An alternative to the complete oxidation of the carbon skeletons to CO
2 is the use of these carbon skeletons for the formation of fat and carbohydrate. Fat is formed from elongation of acetyl units, and so amino acids whose carbon skeletons degrade to acetyl-CoA and ketones may alternatively be used for synthesis of fatty acids. Glucose is split in glycolysis to pyruvate, and pyruvate is the immediate product of
alanine. Pyruvate may be converted back to glucose by elongation to oxaloacetate. Amino acids whose degradation pathways go toward formation of pyruvate, oxaloacetate, or α-ketoglutarate may be used for synthesis of glucose. Therefore, the degradation pathways of many amino acids can be partitioned into two groups with respect to the disposal of their carbon: amino acids whose carbon skeleton may be used for synthesis of glucose (gluconeogenic amino acids) or those whose carbon skeletons degrade for potential use for fatty acid synthesis.
The amino acids that degrade directly to the primary gluconeogenic and TCA cycle precursors, pyruvate, oxaloacetate, and α-ketoglutarate, do so by rapid and reversible transamination reactions:
L-glutamate + oxaloacetate ↔ α-ketoglutarate + L-aspartate
by the enzyme aspartate aminotransferase, which, of course, also can be
L-aspartate + α-ketoglutarate ↔ oxaloacetate + L-glutamate
and
L-alanine + α-ketoglutarate ↔ pyruvate + L-glutamate
by the enzyme alanine aminotransferase. What is quickly apparent is that the amino-N of these three amino acids may be rapidly exchanged and each amino acid rapidly converted to and from a primary compound of gluconeogenesis and the TCA cycle. As described later, compartmentation among different organ pools is the only limiting factor for complete and rapid exchange of the N of these amino acids.
The IDAAs leucine, isoleucine, and valine are grouped together under the heading of the BCAAs because the first two steps in their degradation pathway are common to all three amino acids:
The reversible transamination to keto acids is followed by irreversible decarboxylation of the carboxyl group to liberate CO2. The BCAAs are the only IDAAs that undergo transamination and therefore are unique among IDAAs.
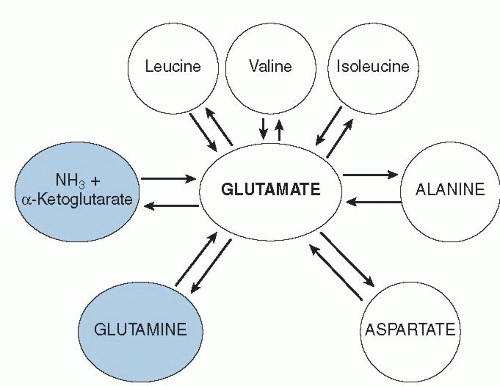
Together, the BCAAs, alanine, aspartate, and glutamate make up the pool of amino-N that can move among amino acids via reversible transamination. As shown in
Figure 1.2, glutamic acid is central to the transamination process. In addition, N can leave the transaminating pool by removal of the glutamate N via glutamate dehydrogenase, or it can enter by the reverse process. The amino acid glutamine is intimately tied to glutamate as well: all glutamine is made from amidation of glutamate, and glutamine is degraded by removal of the amide-N to form NH
3 and glutamate.
A similar process occurs for formation and degradation of asparagine from aspartate. In terms of N metabolism,
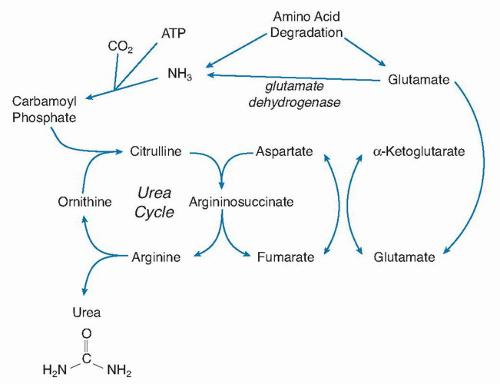
Figure 1.2 shows that the center of N flow in the body is through glutamate. This role becomes even clearer when we look at how urea is synthesized in the liver. The inputs into the urea cycle are a CO
2, adenosine triphosphate (ATP), and NH
3 to form carbamoyl phosphate, which condenses with ornithine to form citrulline (
Fig. 1.3). The second N enters via aspartate to form argininosuccinate, which is then cleaved into arginine and fumarate. The arginine is hydrolyzed by arginase to ornithine, thus liberating urea. The resulting ornithine can reenter the urea cycle. As mentioned briefly later, some amino acids may liberate NH
3 directly (e.g., glutamine, asparagine, and glycine), but most transfer through glutamate first, which is then degraded to α-ketoglutarate and NH
3. The pool of aspartate is small in the body, and aspartate cannot be the primary transporter of the second N into urea synthesis. Rather, aspartate must act like arginine and ornithine as a vehicle for the introduction of the second N. If so, the second N is delivered by transamination via glutamate, again placing glutamate at another integral point in the degradative disposal of amino acid N.
An outline of the degradative pathways of the various amino acids is presented in
Table 1.6. Rather than show individual reaction steps, the major pathways for degradation, including the primary end products, are presented. The individual steps may be found in current textbooks of biochemistry or in older reviews on the subject (
13). Because of the importance of transamination, the majority of the N from amino acid degradation appears via N transfer to α-ketoglutarate to form glutamate. In some cases, the aminotransferase catalyzes the transamination reaction with glutamate bidirectionally, as indicated in
Figure 1.2, and these enzymes are distributed in multiple tissues. In other cases, the transamination reactions are liver specific, are compartmentalized, and act specifically to degrade N, not reversibly exchange it. For example, when leucine labeled with the stable isotope tracer
15N was infused into dogs for 9 hours, considerable amounts of the
15N tracer were found in circulating glutamine + glutamate, alanine, the other two BCAAs, but not in tyrosine (
14)—a finding indicating that the transamination of tyrosine did not proceed backward.
Another reason that the entries in
Table 1.6 do not show individual steps is that the specific pathways of the metabolism of all the amino acids are not clearly defined. For example, two pathways for cysteine are shown. Both are active, but how much cysteine is metabolized by which pathway is not as clear. Methionine is metabolized by conversion to homocysteine. The homocysteine is not directly converted to cysteine; rather, homocysteine condenses with a serine to form cystathionine, which is then broken apart to liberate cysteine, NH
3, and ketobutyrate. The original methionine molecule appears as NH
3 and ketobutyrate, however. The cysteine carbon skeleton comes from the serine. So the entry in
Table 1.6 shows methionine degraded to NH
3, yet this degradation pathway is the major synthesis pathway for cysteine. Because of the importance of the sulfur-containing amino acids, a more extensive discussion of the metabolic pathways of these amino acids may be found in a later chapter.
Glycine is degraded by more than one possible pathway, depending on the text used for reference. The primary pathway, however, appears to be the glycine cleavage enzyme system that breaks glycine into CO
2 and NH
3 and transfers a methylene group to tetrahydrofolate (
15). This pathway has been shown to be the prominent pathway in rat liver and in other vertebrate species (
16). Although this reaction degrades glycine, its importance is the production of a methylene group that can be used in other metabolic reactions.
Synthesis of Dispensable Amino Acids
The IDAAs are those amino acids that cannot be synthesized in sufficient amounts in the body and therefore must be in the diet in sufficient amounts to meet the body’s needs. Therefore, discussion of amino acid synthesis applies only to the dispensable amino acids. Dispensable amino acid synthesis falls into two groups: (a) amino acids that are synthesized by transferring an N to a carbon skeleton precursor that has come from the TCA cycle or from glycolysis of glucose and (b) amino acids that are synthesized specifically from other amino acids. Because this latter group of amino acids depends on the availability of other, specific amino acids, these amino acids are particularly vulnerable to becoming indispensable if the dietary supply of a precursor amino acid becomes limiting. In contrast, the former group is rarely rate limited in synthesis because of the ample precursor availability of carbon skeletons from the TCA cycle and from the labile amino-N pool of transaminating amino acids.
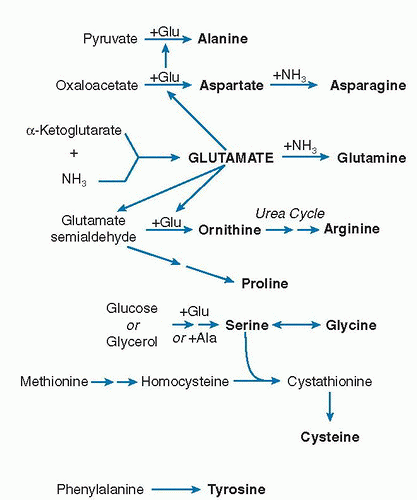
The pathways of dispensable amino acid synthesis are shown in
Figure 1.4. As with amino acid degradation, glutamate is central to the synthesis of several amino acids by providing the N for synthesis. Glutamate, alanine, and aspartate may share amino-N transaminating back and forth among them (see
Fig. 1.2). As
Figure 1.4 is drawn, glutamate derives its N from NH
3 with α-ketoglutarate, and that glutamate goes on to promote the synthesis of other amino acids. Kitagiri and Nakamura (
17) argued that we have little capacity to form glutamate from NH
3 and that the primary source of glutamate N comes from other amino acids via transamination. These amino acids ultimately result from dietary protein intake. Under circumstances of adequate dietary intake, the transaminating amino acids shown in
Figure 1.2 supply more than adequate amino-N to glutamate. The transaminating amino acids act to provide a buffer pool of N that can absorb an increase in N from increased degradation or supply N when there is a drain. From this pool, glutamate provides material to maintain synthesis of ornithine and proline, of which proline is particularly important in protein synthesis of collagen and related proteins.
Serine is produced from 3-phosphoglycerate that comes from glycolysis of glucose. Serine may then be used to produce glycine through a process that transfers a methylene group to tetrahydrofolate. This pathway is listed in
Table 1.6 as a degradative pathway for serine, but it is also a source of glycine and one-carbon unit generation (
15,
16). Conversely, this pathway actively operates backward to form serine from glycine in humans. When [
15N]glycine is given orally, the primary transfer of
15N is to serine (
18). Therefore, significant reverse synthesis of serine from glycine occurs. The other major place where
15N appears was in glutamate and glutamine, a finding indicating that the NH
3 released by glycine oxidation is immediately picked up and incorporated into glutamate and the transaminating N-pool via glutamate dehydrogenase.
All the amino acids shown in
Figure 1.4 have
active routes of synthesis in the body (
13), in contrast to the IDAAs for which no routes of synthesis exist in humans. This statement should be a simple definition of “indispensable” versus “dispensable.” In nutrition, however, we define a dispensable amino acid as an amino acid that is
dispensable from the diet (
3). This definition is different from defining the presence or absence of enzymatic pathways for an amino acid’s synthesis. For example, two of the dispensable amino acids depend on the degradation of IDAAs for their production: cysteine and tyrosine. Although serine provides the carbon skeleton and amino group of cysteine, methionine provides the sulfur through condensation of homocysteine and serine to form cystathionine (
19). From the foregoing discussion, neither the carbon skeleton nor the amino group of serine is likely to be in short supply, but provision of sulfur from methionine may become limiting. Therefore, cysteine synthesis depends heavily on the availability of the IDAA methionine. The same is also true for tyrosine. Tyrosine is produced by the hydroxylation of phenylalanine, which is also
the degradative pathway of phenylalanine. The availability of tyrosine strictly depends on the availability of phenylalanine and the liver’s ability to perform the hydroxylation.
Incorporation of Amino Acids into Other Compounds
Table 1.7 lists some of the compounds that amino acids are converted directly into or are used as important parts of the synthesis of other compounds in the body. The list is not inclusive, and it is meant to highlight important compounds in the body that depend on amino acids for their synthesis. Other important uses of amino acids are for the synthesis of taurine (
20,
21) that is the “amino acid-like” 2-aminoethanesulfonate, found in far higher concentrations inside skeletal muscle than any amino acid (
7). Another important, sulfur-containing compound is glutathione (
22,
23,
24), a tripeptide composed of glycine, cysteine, and glutamate.
Carnitine (
25) is important in the transport of longchain fatty acids across the mitochondrial membrane before fatty acids can be oxidized and is synthesized from ε-N,N,N-trimethyllysine (TML) (
26). TML synthesis occurs from posttranslational methylation of specific lysine residues in specific proteins. TML is liberated when the proteins containing it are broken down (
26). TML can also arise from hydrolysis of ingested meats. In contrast to 3-methylhistidine, TML can be found in proteins of both muscle and other organs such as liver (
27). In rat muscle, TML is approximately one eighth as abundant as 3-methylhistidine.
Amino acids are the precursors for a variety of neurotransmitters that contain N. Glutamate may be an exception in that it serves both as a precursor for neurotransmitter production and is itself a primary neurotransmitter (
28). Glutamate appears important in numerous neurodegenerative diseases from amyotrophic lateral sclerosis to Alzheimer disease. (
29). Tyrosine is the precursor for catecholamine synthesis. Tryptophan is the precursor for serotonin synthesis. Various studies have reported the importance of plasma concentrations of these and other amino acids on the synthesis of their neurotransmitter products. The most common putative relationship cited is the administration of tryptophan, thus increasing brain serotonin levels.
Creatine and Creatinine
Most of the creatine in the body is found in muscle, where it exists primarily as creatine phosphate (
30). When muscular work is performed, creatine phosphate provides the energy through hydrolysis of its “high-energy” phosphate bond that forms creatine with transfer of the phosphate to create an ATP. The reaction is reversible and is mediated
by the enzyme ATP-creatine transphosphorylase (also known as creatine phosphokinase).
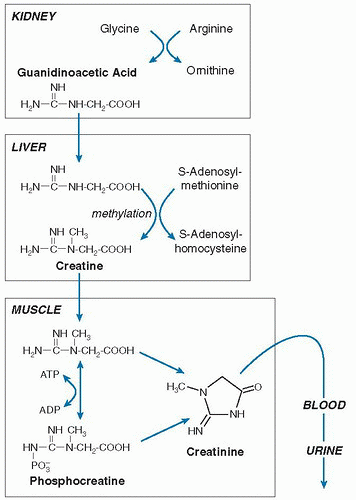
The original pathway of creatine synthesis from amino acid precursors was defined by Bloch and Schoenheimer in an elegant series of experiments using
15N-labeled compounds (
31). Creatine is synthesized outside muscle in a two-step process (
Fig. 1.5). The first step occurs in the kidney and involves the transfer of guanidino group of arginine onto the amino group of glycine to form ornithine and guanidinoacetate. Methylation of the guanidinoacetate occurs in the liver via S-adenosylmethionine to create creatine. Although glycine donates a N and carbon backbone to creatine, arginine must be available to provide the guanidino group, as well as methionine for donation of the methyl group. Creatine is then transferred to muscle, where creatine is phosphorylated. When creatine phosphate is hydrolyzed in muscle to form creatine, most of the creatine is recycled back to the phosphate form. A nonenzymatic process forming creatinine continually dehydrates some of the muscle creatine pool, however. Creatinine is not retained by muscle, but it is released into body water, is then removed by the kidney from blood, and is excreted into urine (
32).
The daily rate of creatinine formation is remarkably constant (≈1.7% of the total creatine pool per day) and depends on the mass of the creatine/creatine-phosphate pool, which is proportional to muscle mass (
33). Thus, daily urinary output of creatinine has been used as a measure of total muscle mass in the body. Urinary creatinine excretion increases within a couple days after a creatine load has been added to the diet, and several more days are required after removal of creatine from the diet before urinary creatinine excretion returns to baseline—a finding indicating that creatine in the diet itself affects creatinine production (
34). Therefore, consumption of creatine and creatinine in meat-containing foods increases urinary creatinine measurements. Although urinary creatinine measurements have been used primarily to estimate the adequacy of 24-hour urine collections, with adequate control of food composition and intake, creatinine excretion measurements are useful indices of body muscle mass (
35,
36).
Purine and Pyrimidine Biosynthesis
The purines (adenine and guanine) and the pyrimidines (uracil, cytosine, and thymine) form the building blocks of DNA and RNA. Purines are heterocyclic double-ring compounds that require incorporation of two glutamine molecules (donation of the amide-N), a glycine molecule, a methylene group from tetrahydrofolate, and the amino-N of aspartic acid for their synthesis as inosine monophosphate. Adenine and guanine are formed from inosine monophosphate by the addition of another glutamine amide-N or aspartate amino-N.
Pyrimidines are synthesized after an amide-N of glutamine is condensed with a CO
2 to form carbamoyl phosphate, which is further condensed with aspartic acid to make orotic acid, the pyrimidine’s heterocyclic 6-member ring. The enzyme that forms this carbamoyl phosphate is present in many tissues for pyrimidine synthesis, but it is not the enzyme found in the liver that makes urea (see
Fig. 1.3). A block in the urea cycle causing a lack of adequate amounts of arginine to prime urea synthesis cycle in the liver, however, will result in diversion of unused carbamoyl phosphate to orotic acid and pyrimidine synthesis (
37). Uracil is synthesized from orotic acid, and cytosine is synthesized from uracil by adding an amide group of glutamine to uridine triphosphate to form cytidine triphosphate.