when it was combined with an “intrinsic factor” (IF) in gastric juice (4). This demonstration sealed the longsuspected connection of PA with achylia gastrica. The third critical achievement was the identification of cobalamin as the extrinsic factor. The synthesis of cobalamin (5, 6) was accompanied by elucidation of its structure by Hodgkin (7), who was also awarded a Nobel Prize for her crystallographic work.
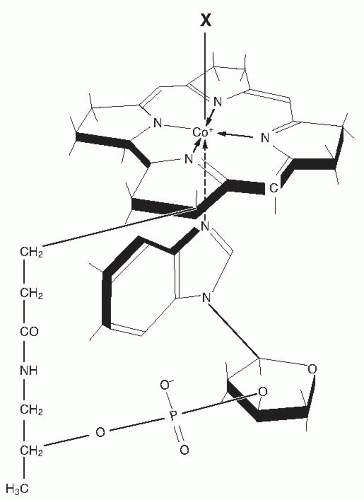 Fig. 27.1. The structure of cobalamin. Attached to the central cobalt atom of the corrin tetrapyrrole and to one of the pyrrole rings is the α-ligand, the 5,6-dimethylbenzimidazole nucleotide, extending below the corrin plane. The β-ligand (marked as X in the figure) above the plane can be any of several moieties such as methyl, 5′-deoxyadenosyl, hydroxyl, or cyanide. (Reprinted with permission from Carmel R. Megaloblastic anemias: disorders of impaired DNA synthesis. In: Greer JP, Foerster J, Lukens JL et al, eds. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia: Lippincott Williams & Wilkins, 2004.) |
as cobalamin and holotranscobalamin (holo-TC) II assays; and measures of functional metabolic status, such as the metabolite biomarkers, methylmalonic acid (MMA) and homocysteine, or complex cellular metabolic indicators such as the deoxyuridine suppression test. When clinical signs of deficiency are evident, one test usually suffices as confirmation (15), but in research and epidemiologic surveys, the frequent absence of clinical identifiers usually requires the application of more than one diagnostic biomarker (16). Unfortunately, no diagnostic gold standard exists.
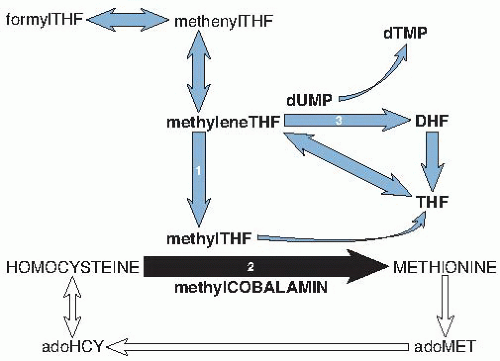 Fig. 27.2. Schematic diagram of the intersection of cobalamin (black arrow) with folate metabolism (blue arrows) and the methionine cycle (white arrows). The direct role of folate in thymidylate synthesis (reaction 3) is also shown. Reaction 1: Reduction of 5,10-methylene tetrahydrofolic acid (THF) to 5-methylTHF by methyleneTHF reductase, which requires riboflavin. Reaction 2: Remethylation of homocysteine to methionine by methionine synthase, with methylTHF and methylcobalamin as cofactors; the THF that is produced is reused in the folate metabolic cycle. Reaction 3: Conversion of deoxyuridylate (dUMP) to deoxythymidylate (dTMP) by thymidine synthase, in which 5,10-methyleneTHF is converted to dihydrofolic acid (DHF). (See the chapter on folic acid for fuller details of folate metabolism.) adoHCY, S-adenosylhomocysteine; adoMET, S-adenosylmethionine. |
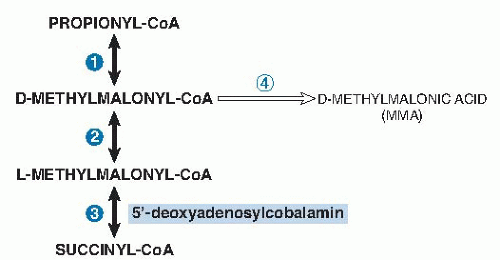 Fig. 27.3. The sole function of 5′-deoxyadenosylcobalamin in humans. The mitochondrial conversion of propionyl-coenzyme A (CoA), which derives from diverse sources to succinyl-CoA, which enters the tricarboxylic acid cycle, passes through three reversible reactions. Reaction 1: Carboxylation of propionyl-CoA by propionyl-CoA carboxylase, requiring adenosine triphosphate (ATP), biotin, and magnesium. Reaction 2: Racemization of D-methylmalonyl-CoA by methylmalonyl-CoA racemase. Reaction 3: Intramolecular rearrangement of L-methylmalonyl-CoA to succinyl-CoA by L-methylmalonyl-CoA mutase, which requires 5′-deoxyadenosylcobalamin. In addition, an irreversible side reaction converting D-methylmalonyl-CoA to methylmalonic acid, mediated by D-methylmalonyl-CoA hydrolase, produces methylmalonic acid (reaction 4). The metabolic fate of methylmalonic acid is largely unknown, but a fraction is excreted by the kidneys. |
missed (28). This change, adopted by many laboratories, instantly increased the frequency of diagnosed deficiency. The overdiagnosis it creates is substantial. For example, the 5.3% to 24.8% frequency of “abnormal” cobalamin values in four surveys became 40.5% to 71.7% in the unexceptional elderly populations (15). More relevantly, reanalysis showed that only one third of the persons thus recategorized had MMA or homocysteine abnormalities, thus, making two thirds of the new diagnoses metabolically suspect; moreover, very few of the one third had clinical deficiency (10, 16). Furthermore, the advantages of overdiagnosis remain unrealized because the health risks of SCCD and the benefits of intervention in such cases remain unproven.
TABLE 27.1 CONDITIONS FREQUENTLY ASSOCIATED WITH LOW SERUM COBALAMIN LEVELS | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
unclear what mild MMA elevations without any other abnormalities mean (16, 51, 52). The improvement of many isolated MMA elevations after cobalamin is given, which suggests that these elevations represented mild SCCD (23, 30, 51). However, normal MMA levels often decline after cobalamin therapy, too—a finding suggesting regression to the mean or supersaturation of methylmalonyl-CoA mutase by the cobalamin as alternative explanations. A noteworthy longitudinal study of 432 patients left untreated over 1 to 3.9 years observed that 44% of isolated mild elevations of MMA improved spontaneously and only 16% progressed (53). Antibiotics sometimes reduce cobalamin-unresponsive MMA elevation (23, 54)—a finding suggesting that increased propionate metabolism by some intestinal bacteria may directly elevate MMA without cobalamin deficiency.
in milk (65). Plants are negligible sources of cobalamin, although dried green and purple algae contain cobalamin that may be bioavailable (65). Better methods are needed to quantitate food content and to differentiate cobalamins that are usable by humans from corrinoids that are not (65). As important as content are features such as bioavailability, which can vary 10-fold among different foods and stability after cooking or processing. Milk and cobalamin-fortified cereals are particularly efficient sources in the US diet and fish in the Norwegian diet, and all exceed meat in their cobalamin bioavailability (65, 66, 67).
TABLE 27.2 COBALAMIN BIOAVAILABILITY FROM A SINGLE ORAL DOSE GIVEN TO PERSONS WITH (A) NORMAL ABSORPTION AND (B) ABNORMAL ABSORPTION WITHOUT INTRINSIC FACTOR (PERNICIOUS ANEMIA)a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
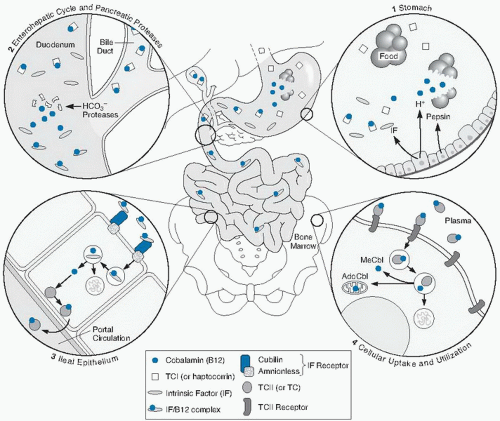 Fig. 27.4. Absorption and cellular uptake cycle of cobalamin in humans: 1, secretion of gastric intrinsic factor (IF), acid, and pepsin and release of cobalamin from food and binding to TC I (R binder or haptocorrin); 2, biliary and pancreatic secretion and degradation of TC I by pancreatic enzymes; 3, ileal cell uptake of IF-cobalamin by cubam (the cubilin-amnionless receptor), lysosomal processing, and transfer of transcobalamin (TC) II-cobalamin to the portal circulation; 4, cellular uptake (e.g., in bone marrow) of plasma TC II-cobalamin, lysosomal processing, and release of cobalamin for mitochondrial or cytoplasmic attachment to enzymes. AdoCbl, adenosylcobalamin; MeCbl, methylcobalamin. (Modified from Carmel R, Rosenblatt, DS. Disorders of cobalamin and folate metabolism. In: Handin RI, Lux SE, Stossel TP, eds. Blood: Principles and Practice of Hematology. 2nd ed. Philadelphia: Lippincott Williams & Wilkins, 2003. Originally adapted with permission from Carmel R. Cobalamin deficiency. In: Carmel R, Jacobsen DW, eds. Homocysteine in Health and Disease. Cambridge: Cambridge University Press, 2001.) |
cobalamin reaches the bloodstream, its transport and uptake depend on TC II. TC I also binds cobalamin in the blood, but its role appears to involve withholding cobalamin and, possibly more importantly, nonfunctional corrinoid analogs from cells. Hepatic clearance of cobalamin through bile approximates 1.4 µg daily; approximately 70% is normally reabsorbed, presumably by IF, whereas the remainder is lost in feces (13) along with most corrinoid analogs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


